Artikli meditsiiniline ekspert

Uued väljaanded
Arachnodactyly
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
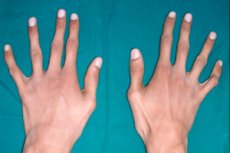
Üks haruldastest pärilikest sidekoe patoloogiatest on arahnodaktüülia – sõrmede deformatsioon, millega kaasneb torukujuliste luude pikenemine, skeleti kõverused, südame-veresoonkonna ja nägemisorganite häired.
Haigust kirjeldasid esmakordselt dr Williams ja prantsuse lastearst Marfan, kes nimetasid seda dolihhostenomeliaks. Hiljem ilmus maailmameditsiinis termin Marfani sündroom ja alates 1902. aastast on kasutatud nime arahnodaktüülia. Patoloogia on seotud kaasasündinud sidekoe häiretega ja üheks juhtivaks tunnuseks peetakse piklikke, hõrenenud ja kõverdunud "ämbliku" sõrmi. [ 1 ]
Epidemioloogia
Arahnodaktüüliat peetakse pärilikuks sidekoe monogeenseks patoloogiaks, millel on autosomaalne dominantne pärandumine, kõrge penetrance ja erineva ekspressiivsusega.
Ligikaudu 75–80% juhtudest on haigus pärilik ja ülejäänud juhtudel tekib see spontaansete mutatsioonide tagajärjel (eelkõige punkt-missense mutatsioonide tõttu). [ 2 ]
On tõestatud, et arahnodaktüülia patogeneetilised tunnused on põhjustatud muutustest mitmes geenis (95% juhtudest):
- fibrilliin-1 geenis (on uuritud umbes tuhat mutatsiooni) kromosoomis 15q21.1;
- TGFβR1 või TGFβR2 geen 9. kromosoomis ja 3p24.2-P25.
Need muutused mõjutavad otseselt kliiniliste ilmingute ulatust.
I tüüpi kollageeni α-2 ahela mutatsioone on kirjeldatud 5%-l patsientidest.
Arahnodaktüülia levimus on umbes 2 juhtu 5000 inimese kohta. Rassilist ega soolist määratlust ei ole. [ 3 ]
Põhjused arachnodaktia
Arahnodaktüülia tekib spontaanse mutatsiooni tagajärjel. Haigusele ei ole iseloomulikud kaasasündinud pärilike patoloogiate üldtuntud tunnused ning piklikud sõrmed avalduvad peamiselt Marfani sündroomi ja homotsüstinuuria korral.
See haigus on haruldane ja kõigil leitud patsientidel oli FBN1 geeni (fibrilliini geen) mutatsioon, mis paikneb 15. kromosoomis q21.1 piirkonnas. Sündroomi kliinilist polümorfismi saab seletada mutatsioonide massiga, millest umbes 15% on mutatsioonilised muutused, mis tekkisid viljastumise ajal. [ 4 ]
Arahnodaktüülia atüüpilised vormid on põhjustatud mutatsioonidest teistes geenides: on avastatud seos patoloogia arengu ja LTBP3, R geeni mutatsioonide vahel, mis lokaliseerub 14. kromosoomis q24 piirkonnas ja transformeerib kasvufaktorit.
Valdav enamus mutatsioonidest on nn missense-mutatsioonid (umbes 57%). Üle 20% on väikesed deletsioonid, millega kaasnevad kromosoomiosade kadu, umbes 12% on splaissingu saidi mutatsioonid, 8% on nonsenssmutatsioonid ja 2% on ulatuslikud ümberkorraldused ja deletsioonid. [ 5 ]
Kaasasündinud kontrakturaalne arahnodaktüülia on põhjustatud valgu fibrilliin-2 sünteesihäirest. Probleemi põhjustab mutatsioon FBN2 geenis, mis asub viiendas kromosoomis q23-q31 piirkonnas. Kõik mutatsioonid on valdavalt punktmutatsioonid, mis lülitavad koodoni kodeerima mõnda teist aminohapet. [ 6 ]
Homotsüstiinuuria tekib asendamatu aminohappe metioniini metabolismi häire taustal. Patoloogia aluseks võib olla järgmiste ensüümide aktiivsuse kadu või selle vähenemine:
- Tsüstatsiiniini beeta-süntaasi (CBS) ensüüm. Patoloogia on B6 resistentne ja avaldub B6-sõltumatule vormile iseloomulike tunnustega , kuidB6- vitamiini preparaadi kasutuselevõtt ei vii paranemiseni.
- Ensüüm N5, N10 metüleentetrahüdrofolaadi reduktaas (MTHFR). See ensümaatiline aine on substraat, mis põhjustab homotsüsteiini demetüleerimist ja selle muundumist metioniiniks, mis on paljude valkude ja peptiidide koostisosa.
- Ensüüm N5 metüleentetrahüdrofolaat. Jutt käib B6 -vitamiinist sõltuvast patoloogiast, mis tekib B6-vitamiini puuduse korral . Vitamiinipreparaadi manustamine organismi parandab patsiendi seisundit.
- Homotsüsteiini transmetülaasi ensüüm. Tekib kobalamiini – B12 -vitamiini – ainevahetushäirete taustal.
Riskitegurid
Arahnodaktüülia on geneetiliselt määratud haigus, mida iseloomustab sidekoe süsteemne kahjustus. Patoloogia on seotud muutustega fibrilliin 1 geenis, mis lokaliseerub 15. kromosoomi lühikesel harul lookuses 21.1.
Arahnodaktüülia pärandub autosomaalselt dominantsel viisil, kõrge penetrantsuse ja varieeruva ekspressiivsusega. Patoloogia võib avalduda nii mees- kui ka naissoost järglastel.
Pathogenesis
Üle 50% inimese kehamassist moodustavad sidekoe struktuurid, eelkõige skelett, nahk, veresoonte võrgustik, lümf ja veri.
Sidekoe rakke esindavad fibroblastid ja nende alatüübid: osteoblastid, kondrotsüüdid, keratoblastid, odontoblastid, samuti makrofaagid ja nuumrakud.
Embrüonaalne sidekude on materjal konstitutsiooniliste, geneetiliste ja epigeneetiliste komponentide moodustumiseks. Sidekoehaigused kajastuvad ühel või teisel määral kogu kehas tervikuna, selle funktsionaalses võimekuses ja konstitutsioonis.
Arahnodaktüülia korral toimub geenis nukleotiidide asendus, mis sisaldab struktuurilist teavet fibrilliin-1 peptiidi kohta. See valguaine kuulub glükoproteiinirühma, osaleb mikrofibrillaarses süsteemis ja moodustab sidekoe elastsete fibrillide aluse.
Kude säilitab stabiilse struktuuri tänu rakkudevahelisele maatriksile, mis sisaldab palju kasvufaktoreid, mis tagavad süstemaatilise rakkude uuenemise. Suured veresooned ja sidemed sisaldavad arvukalt elastiini fibrille, mille kahjustus põhjustab arahnodaktüülia peamisi sümptomeid.
Transformeeriv kasvufaktor β on oluliselt kahjustatud: selle inaktiivne vorm ei ole seotud, mis viib selle faktori bioloogilise aktiivsuse suurenemiseni.
Fibrillaarsed häired põhjustavad kiudude ebanormaalset moodustumist, mille tulemuseks on naha ja teiste sidekoe struktuuride tugevuse ja elastsuse kaotus.
Kui kollageeni struktuur on häiritud, on häiritud hemostaasi peamine lüli.
Eksperdid märgivad hormonaalse tasakaalutuse seotust sidekoe struktuuride, sealhulgas arahnodaktüülia, anomaaliate tekke ja progresseerumisega. Trombootilised sümptomid on määratud reoloogiliste häiretega - eelkõige vere viskoossuse muutustega brachiotsefaalse piirkonna modifitseeritud anumates.
Seedetrakt on mõjutatud kollageenihäirete tõttu: täheldatakse hüpomotoorset sapiteede düskineesiat, diafragma söögitoru avause herniat, hepatobiliaarseid anomaaliaid, kroonilist "kustutatud" gastroduodeniidi vormi, dolihosigmat.
Kuidas arahnodaktüülia pärandub?
Arahnodaktüülia pärandub autosomaalselt dominantselt, aga võib esineda ka spontaansete mutatsioonide tagajärjel. See tähendab, et patoloogia ei pea tingimata avalduma ühel lähisugulasel – näiteks vanematel, vanavanematel. Patoloogia aluseks olevad mutatsioonid võivad endast märku anda juhuslikult.
Sümptomid arachnodaktia
Meditsiinis eristatakse latentset ja avalikku arahnodaktüüliat. Latentse arahnodaktüülia korral muutused esinevad, kuid need on isoleeritud ja piirduvad ühe või kahe organsüsteemiga. Avatud arahnodaktüülia avaldub mitmete nähtavate häiretena, millel on erineva intensiivsusega sümptomid. Sellisel juhul võib patsiendi seisund olla aastakümneid suhteliselt stabiilne või pidevalt progresseeruda, avaldudes seoses teiste organite ja süsteemidega.
Sageli saab arahnodaktüülia sündroomi tuvastada esimestel päevadel pärast lapse sündi.
Arahnodaktüülia peamine väline tunnus on õhukesed ja piklikud sõrmed, mis lühikeste kõõluste tõttu omandavad tüüpilise kumeruse ja meenutavad ämblikujalgu.
Ämbliksõrmed ehk arahnodaktüülia muutuvad selgelt nähtavaks peaaegu vastsündinust alates. Kuid märk muutub ilmsemaks umbes kolmeaastaselt.
Iseloomulikud on ka kasvunäitajad: lapsed on tavaliselt pikad, käsivarred ja sääred on pikad, jäsemed ebaproportsionaalselt suured ja kõhnad. Ebaproportsionaalsus on eriti ilmne vöökoha ja põlvekederde kõrge lokaliseerimise korral. [ 7 ]
Muud skeletihaigused hõlmavad järgmist:
- kitsenenud ja piklik kolju kuju (dolichocephalic) ebanormaalselt moodustunud näopiirkonnaga;
- lehtrikujuline rindkere konfiguratsioon (nn "lind");
- kõver selg;
- kaasasündinud puusaliigese nihestused;
- harjumuspärased põlve-õla nihestused;
- kanna luu kõverus;
- osteofüütide moodustised – luukasvajad;
- lamedad jalad;
- rasvakihi ebapiisav areng.
Arahnodaktilia täiendavateks sümptomiteks võivad olla:
- lühinägelikkus;
- sinakas kõvakest;
- läätse subluksatsioon;
- südame ja veresoonte häired (südamerikked, aordi aneurüsm);
- väljendunud kõhnus;
- liigeste hüpermobiilsus;
- "kaarjas" taevas.
Arahnodaktüüliat iseloomustavad piklikud torukujulised luud, mis põhjustavad mitmesuguseid skeleti kõverusi. Lisaks on paljudel patsientidel normaalse vaimse arengu taustal püramiidsed häired, kõõluste reflekside asümmeetria, anosokoria ja nüstagm.
Kui me räägime arahnodaktiilist haigusest, millega kaasneb homotsüstinuuria, võib patsiendil esineda järgmised sümptomid:
- perioodilised krampide rünnakud;
- sekundaarne glaukoom koos läätse subluksatsiooniga;
- võrkkesta irdumine, astigmatism;
- arteriaalsete tüvede (sh neerude, aju) kahjustus;
- osteoporoos;
- tromboos;
- vaimne alaareng.
Arahnodaktüülia vastsündinutel
Enamasti saab arahnodaktüüliat lapsel tuvastada juba esimestel elupäevadel. Vanusega kipuvad sümptomid aga süvenema.
Esimesed märgid, mis võimaldavad kahtlustada haigust, on tavaliselt järgmised:
- ebatavaliselt pikad jäsemed ja sõrmed;
- lapse alakaal piisavas füüsilises seisundis;
- piklik, pikk nägu;
- keharasva puudumine, halvasti arenenud lihased;
- liigne liigeste paindlikkus.
Umbes nelja-aastasest east, arahnodaktüülia taustal, hakkab rindkere muutuma, selgroog muutub kõverdunuks ja tekivad lamedad jalad.
Nägemisorganite osas täheldatakse lühinägelikkust, läätse ektoopiat, sarvkesta konfiguratsiooni muutumist, strabismust, iirise ja võrkkesta hüpoplastilisi protsesse. Selliseid häireid võib märgata juba lapse esimese 2-3 eluaasta jooksul, kuigi need progresseeruvad aastate jooksul.
Suurim oht on südame-veresoonkonna häired. Kui sellised häired esinevad, on sobiva ravi puudumisel suur risk patsiendi surmaks juba lapsepõlves. Eriti ohtlike patoloogiate hulgas võib välja tuua veresoonte seinte kahjustused, südame väärarengud ja pärgarterite defektid. Mõnikord avastatakse juba esimesel eluaastal lastel süvenev südamepuudulikkus.
Arahnodaktüüliaga võivad kaasneda ka teiste organite, eriti närvisüsteemi, bronhide ja kopsude, naha ja kuseteede kahjustused. Sellised häired avastatakse diagnostiliste protseduuride käigus.
Tüsistused ja tagajärjed
Arahnodaktüülia kõige sagedasemad kõrvaltoimed on südame ja hingamisteede kahjustused. Lisaks ilmnevad nägemisprobleemid kuni nägemise kadumiseni.
Arahnodaktüüliaga patsiendid vajavad kõigi olemasolevate patoloogiate kompleksset ravi. Kui sellist ravi ei teostata, väheneb patsientide eluiga märkimisväärselt: harvad patsiendid suudavad ilma meditsiinilise sekkumiseta elada 40-aastaseks. Aastate jooksul süvenevad kardiovaskulaarsüsteemi probleemid ja suureneb ulatuslike verejooksude oht traagilise tulemusega.
Arahnodaktüülia all kannatavad inimesed peaksid regulaarselt läbima uuringuid ja ravima kaasnevaid patoloogiaid: ainult sellisel juhul on neil kõik võimalused elada pikka elu ilma tõsiste tüsistusteta.
Marfani sündroom ja arahnodaktüülia vajavad erilist tähelepanu, kui naispatsient rasestub. Sellises olukorras on oluline patsienti regulaarselt ja põhjalikult uurida ning tema südame-veresoonkonna seisundit meditsiiniliselt toetada. See on ainus viis tagada edukas rasedus ja lapse turvaline sünd.
Negatiivsete mõjude vältimiseks peaksid arahnodaktüüliaga patsiendid regulaarselt arstide poolt läbi vaatama, et vältida südame-veresoonkonna ja lihasluukonna patoloogiate teket. Neil on soovitatav elada ainult tervislikku eluviisi ja mõõdukat füüsilist aktiivsust.
Diagnostika arachnodaktia
Arahnodaktüülia kliiniline diagnostika hõlmab kaebuste kogumist, perekonnaanamneesi, fenotüübi hindamist ning antropomeetrilist ja füüsilist läbivaatust. Oluline on kindlaks teha sidekoe patoloogiate ilmingute esinemine otsestel sugulastel: selleks viiakse läbi genealoogiline uuring. [ 8 ]
Arahnodaktüülia laboratoorne diagnostika hõlmab teatud tüüpi sidekoe seisundi uurimist, mida esindavad luu- ja kõhrkude, veri ja lümf. Kõige informatiivsemaks peetakse oksüproliini ja glükosaminoglükaanide taseme uuringut päevases uriinimahus. Lisaks hinnatakse lüsiini, proliini ja oksüproliini sisaldust veres. Erinevat tüüpi kollageenide muutunud suhete ja kollageenkiudude struktuuriliste kõrvalekallete kindlakstegemiseks määratakse tüpiseerimine kaudse immunofluorestsentsi meetodil, kasutades kollageeni ja fibronektiini polüklonaalseid antikehi. [ 9 ] Sõltuvalt näidustustest tehakse sidekoe ainevahetusprotsesside kvaliteedi kajastamiseks järgmised testid:
- glükosaminoglükaanid, fibrilliin, fibronektiin;
- hüdroksüproliini, 1. tüüpi kollageeni biosünteesi markerite, 1. tüüpi prokollageeni aminoterminaalsete propeptiidide, 1. tüüpi kollageeni lagunemise markerite, galaktosüüloksülüsiini, deoksüpüridinoliini uuring;
- kollageeni metabolismi regulatsiooni analüüs (maatriksmetalloproteinaaside ja maatriksmetalloproteinaaside koe inhibiitorite, transformeeriva kasvufaktori uuring);
- mikro- ja makroelementide analüüs (kaltsiumi ja magneesiumi, fosfori, raua, väävli ja vase, koobalti ja seleeni, tsingi ja mangaani, fluori ja vanaadiumi, räni ja boori sisaldus);
- Luukoe moodustumise markerite ja ümberehitusprotsesside kiiruse uuring (osteokaltsiini, luu aluselise fosfataasi, paratüreoidhormooni, somatotroopse hormooni, prolaktiini, D3-vitamiini , pentosidiini, homotsüsteiini sisalduse analüüs uriinis ja veres).
Instrumentaalne diagnostika on suunatud olemasolevate arenguhäirete tuvastamisele ja organite ja süsteemide funktsionaalse seisundi hindamisele. Arahnodaktüülia korral esinevad kardiovaskulaarsed häired nõuavad diagnostiliste protseduuride kohustuslikku lisamist järgmistesse diagnostilistesse loenditesse:
- elektrokardiograafia;
- 24-tunnine EKG jälgimine;
- südame ja veresoonte ultraheliuuring.
Vaskulaarseid anomaaliaid uuritakse kompuutertomograafia või magnetresonantstomograafia abil. Täiendavad diagnostilised protseduurid võivad hõlmata järgmist:
- Kõhuõõne organite, urogenitaalsüsteemi ultraheli;
- Rindkere organite ja puusaliigeste röntgen; [ 10 ]
- Selgroo kompuutertomograafia või magnetresonantstomograafia.
Pärast kõiki vajalikke uuringuid suunatakse patsient geneetilisele nõustamisele.
Diferentseeritud diagnoos
Bealsi sündroomi ja Marfani sündroomi arahnodaktüülia vahel on fenotüübiline sarnasus (vastavalt FBN2 ja FBN1 geenide mutatsioonid), mis on tingitud valguainete fibrilliin 1 ja fibrilliin 2 peaaegu täielikust identsusest. Pole üllatav, et Bealsi sündroomi teine nimetus on arahnodaktüülia kontraktuur. [ 11 ]
Diferentseerimist viiakse läbi ka teiste sidekoe patoloogiatega:
- Stickleri sündroom;
- Ehlers-Danlose sündroom;
- homotsüstinuuria;
- artrogrüpoos.
Bealsi sündroomi iseloomustab marfanoidne fenotüüp, suurte ja väikeste liigeste kaasasündinud paindekontraktuurid, randme ja jala arahnodaktüülia, selgroo kõverus ja ebanormaalne kõrva kuju (nn "kortsus" kõrvakesed). Diagnoosi kinnitamiseks tehakse FBN2 geeni molekulaarne analüüs.
Artrogrüpoosi iseloomustavad mitmed liigesekontraktuurid, lühike kasv ja vähenenud intelligentsus. Sõrmede ja kõrvade kuju on tähelepanuväärne. [ 12 ]
Ehlers-Danlose sündroomi iseloomustab küfoskolioos, lehtrikujuline rindkere kõverus, väljendunud lamedad jalad ja halvenenud refraktsioon. Tüüpilised on suurenenud liikuvus ja liigeste nihestused, naha suur venivus ja tundlikkus.
Stickleri sündroomi iseloomustavad liigese mahu ja jäikuse muutused.
Kellega ühendust võtta?
Ravi arachnodaktia
Arahnodaktüülia on geneetiline patoloogia ja praegu puuduvad meditsiinil meetodid geenidefektide korrigeerimiseks. Seetõttu on ravi suunatud patsiendi seisundi optimeerimisele, patoloogia süvenemise ennetamisele ja sümptomaatiliste ilmingute kõrvaldamisele. Määratakse kompleksne ravi, mis hõlmab korraga mitut eriarsti, olenevalt kõige ilmekamate sümptomite tüübist: sageli on vaja lisaks pöörduda ortopeedi, kardioloogi, oftalmoloogi, gastroenteroloogi ja teiste erialade arstide poole. [ 13 ]
Patsientide kliinilise ravi soovituste hulgas peetakse üldpõhimõteteks järgmist:
- füüsilise aktiivsuse piiramine, kuid mitte selle täielik kõrvaldamine (osana südame-veresoonkonna toetamisest);
- ravimteraapia;
- vajadusel – südame ja veresoonte enim kahjustatud piirkondade kirurgiline korrigeerimine;
- ortopeediline korrektsioon;
- spaahooldus, füsioteraapia, terapeutiline võimlemine.
Arahnodaktüüliaga patsientide toitumine peaks sisaldama piisavas koguses kõrge valgusisaldusega tooteid, mis on rikastatud mikroelementide, vitamiinide ja rasvhapetega. Soovitatav on süüa liha, kala, mereande, ube ja pähkleid. Kui arahnodaktüülia on põhjustatud homotsüstinuuriast, siis on loomse valgu tarbimine järsult piiratud.
Saleda ja pika kasvuga lastele soovitatakse juba varases eas oma toidusedelisse lisada puuvillaseemne- ja sojaõli, päevalilleseemneid, searasva ja seapekki. Lisaks pakutakse preparaate, mis sisaldavad polüküllastumata rasvhappeid, näiteks omega-rasvhappeid, mis pärsivad somatotroopse hormooni tootmist.
Valgu ainevahetuse normaliseerimiseks on ette nähtud B-rühma vitamiinid. Neid saab ka toiduainetest: tatar, herned, maks.
On väga oluline, et askorbiinhape siseneks patsiendi kehasse toiduga. Sel eesmärgil on toidus tingimata kaasatud kibuvitsa infusioon, paprikad, kapsas, tsitrusviljad, samuti astelpaju ja porrulauk.
Vajadusel pakutakse patsientidele ortopeedilist korrektsiooni, mis on vajalik selgroo ja liigeste koormuse vähendamiseks.Sel eesmärgil kasutatakse ortopeedilisi jalatseid, põlve- ja muid seadmeid, sisetallasid, elastseid sidemeid.
Kirurgiline ravi viiakse läbi vastavalt näidustustele.
Ravimid
Arahnodaktüülia medikamentoosne ravi viiakse läbi 1-2 korda aastas, olenevalt patsiendi seisundist ja patoloogiliste sümptomite raskusastmest. Ravikuuri kestus määratakse individuaalselt ja on keskmiselt 4 kuud. [ 14 ]
Kollageeni moodustumise stimuleerimiseks on ette nähtud järgmised ravimid: Piaskledin 300, L-lüsiin või L-proliin koos komplekssete multivitamiinipreparaatidega, mis sisaldavad askorbiinhapet, B-vitamiine, tokoferooli, magneesiumi, tsinki, seleeni, vaske.
Kondroprotektorite seas on kõige optimaalsem kasutada kondroitiinsulfaati ja glükoosamiinsulfaati - ravimeid, mis osalevad kondrotsüütide metabolismi reguleerimises, ensüümide sünteesi pärssimises, kondrotsüütide tundlikkuse suurendamises ensümaatilise toime suhtes, anaboolsete protsesside stimuleerimises jne. Kombineeritud aineid peetakse optimaalseteks sellisteks ravimiteks: Teraflex, Arthroflex, Arthra jne.
Mineraalainete ainevahetust stimuleerivad ravimid, mis normaliseerivad fosfori-kaltsiumi protsesse. Valitud ravimiteks on sageli D-vitamiini aktiivsed vormid: Alpha D3 Teva, Oxidevit, Bonviva jne. Samal ajal kasutatakse fosfori-, kaltsiumi- ja magneesiumipreparaate. Ravi ajal kontrollitakse kaltsiumi ja fosfori taset veres või uriinis umbes iga 20 päeva tagant ning tehakse vereanalüüs aluselise fosfataasi määramiseks.
Keha bioenergeetilise seisundi parandamiseks on võimalik välja kirjutada fosfadeni, riboksiini, letsitiini, Elkari, koensüümi Q10.
Ligikaudne raviskeem võib välja näha selline:
- Kondroprotektor vanusele sobivas annuses, koos toidu ja piisava koguse veega. Ühe ravikuuri kestus on 3-4 kuud.
- L-proliini annuses 500 mg (lastele alates 12. eluaastast ja täiskasvanutele) võetakse pool tundi enne sööki 1-2 korda päevas kuue nädala jooksul. Vajadusel võib lisaks määrata aminohapete kompleksi - L-proliini, L-lüsiini, L-leutsiini koguses 10-12 mg/kg kehakaalu kohta. Sissevõtmine toimub 1-2 korda päevas kahe kuu jooksul.
- Vitamiinide ja mineraalide komplekspreparaadid Centrum, Vitrum või Unicap, annuses, mis arvestab vanust. Manustamise kestus on 4 nädalat.
See raviskeem on sobiv, kui arahnodaktüüliaga patsient kurdab lihasluukonna probleemide üle ning testi tulemused näitavad glükosaminoglükaanide suurenenud eritumist igapäevases uriinianalüüsis ja vabade aminohapete taseme langust veres.
Reeglina on patsiendid ravi hästi vastu võtnud, ilma eriliste kõrvaltoimeteta. Kui tuvastatakse ülitundlikkusreaktsioone, asendatakse ravimid ja raviskeemi kohandatakse.
Füsioteraapia
Arahnodaktüülia füsioterapeutilised protseduurid määratakse vastavalt näidustustele. Näiteks ebapiisava osteogeneesi korral, luumurdude paranemise parandamiseks või osteoporoosi tunnuste korral on soovitatav elektroforees 5% kaltsiumkloriidi, 4% magneesiumsulfaadi, 2% vasksulfaadi või 2% tsinksulfaadiga.
Kui avastatakse vegetatiivseid-vaskulaarseid häireid, kasutatakse 1% naatriumkofeiinbensoaati, efedriinvesinikkloriidi või mesatooni.
Neerupealise koore funktsiooni aktiveerimiseks on ette nähtud ravimelektroforees 1,5% etimisooliga ja neerupealise piirkonna detsimeeterravi. [ 15 ]
Veresoonte toonuse stabiliseerimiseks on soovitatavad veeprotseduurid, mis soodustavad veresoonte "võimlemist". Suurepärased on vannid nagu süsinikdioksiid, mänd, kloriid-vesinik, vesiniksulfiid ja radoon. Harjutatakse hõõrumisi, douse'e, kontrastdušše, vahu- ja soolavanne.
Kõhrekoe seisundi parandamiseks kasutatakse magnetravi, induktoteraapiat, laserteraapiat ja elektroforeesi dimetüülsulfoksiidiga (Dimexide).
Kirurgiline ravi
Arahnodaktüülia kirurgilised operatsioonid määratakse rangelt vastavalt näidustustele. Näiteks kui klapiklappide prolapsi taustal avastatakse raskeid hemodünaamilisi häireid, tehakse massiivne aordi aneurüsm, klapivahetus ja kahjustatud aordi segment. [ 16 ]
Kui rindkere tugeva kõveruse tagajärjel tekivad hingamisteede ja kardiovaskulaarsüsteemi väljendunud funktsionaalsed häired, tehakse torakoplastika.
3-4-kraadise raske skolioosiga kaasneva progresseeruva valusündroomi korral on näidustatud ka kirurgiline sekkumine. Oftalmoloogilisest vaatenurgast on läätse eemaldamise absoluutsed näidustused läätse subluksatsioon, mida komplitseerib sekundaarne glaukoom, samuti katarakt ja võrkkesta degeneratiivsed muutused, millega kaasneb suur irdumise oht.
Arahnodaktüülia ja muude sidekoe struktuure mõjutavate häiretega patsientidel tehakse operatsioone ainult suhtelise kliinilise ja biokeemilise leevenduse staadiumis. Pärast sekkumist on kohustuslik pikaajaline meditsiiniline jälgimine ja intensiivravi ravimitega, mis parandavad sidekoe ainevahetust.
Ärahoidmine
Arahnodaktüülia on haruldane geneetiline patoloogia. Mõnikord tekib see spontaanselt pealtnäha tervetel vanematel. Seetõttu on haigust väga-väga raske eelnevalt ennetada.
Kui ühel potentsiaalsetest vanematest on keeruline perekonnaanamnees – see tähendab, et on teada, et keegi perekonnas on kannatanud arahnodaktüülia all –, peaksid abikaasad lapse planeerimise etapis külastama geneetikut ja läbima geneetilise uuringu. Pärast raseduse kinnitamist teeb arst sünnieelse diagnostika, sealhulgas loote ultraheliuuringu ja mitmed biokeemilised testid:
- ema vereanalüüs;
- lootevee analüüs;
- koorionivilla proovide võtmine;
- Platsenta rakkude ja nabaväädivere analüüs.
Arahnodaktüüliaga patsiendid on kogu elu jooksul meditsiinilise jälgimise all. Patsientide ennetusmeetmed on suunatud tüsistuste ennetamisele. Sel eesmärgil tehakse südame kirurgilist korrektsiooni, määratakse ravimravi, et välistada trombi tekke oht. Perioodiliselt manustatakse antibiootikumravi kuure.
Prognoos
Arahnodaktüüliaga patsientide eluprognoosi määrab eelkõige kaasuvate haiguste raskusaste - eelkõige südame-veresoonkonna, lihasluukonna ja nägemisorganite haigused. Kõige sagedamini raskendab patoloogiat aordi rebend ja dissektsioon. Seda tuleb arvesse võtta ja teha õigeaegne südameoperatsiooni korrektsioon, mis aitab pikendada patsiendi eluiga ja parandada selle kvaliteeti.
Kui diagnoos tehti õigeaegselt ja arsti soovituste järgimisel võib prognoosi nimetada tingimuslikult soodsaks. Meditsiiniline tugi leevendab oluliselt arahnodaktüülia kulgu ning patsientidel on võimalus elada normaalset ja suhteliselt täisväärtuslikku elu ilma tõsiste tüsistuste tekkimiseta.