Huntingtoni tõbi
Last reviewed: 25.06.2018

Meil on ranged allikate valiku juhised ja lingime ainult mainekatele meditsiinilistele saitidele, akadeemilistele uurimisasutustele ja võimaluse korral meditsiiniliselt eelretsenseeritud uuringutele. Pange tähele, et sulgudes olevad numbrid ([1], [2] jne) on klõpsatavad lingid nendele uuringutele.
Kui arvate, et mõni meie sisust on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
Huntingtoni tõbi on autosoom-dominantne neurodegeneratiivne häire, mida iseloomustab progresseeruv kognitiivne langus, tahtmatud liigutused ja motoorse koordinatsiooni häired, mis algavad keskeas. Diagnoosi kinnitab geneetiline testimine. Ravi on peamiselt sümptomaatiline. Geneetilisi teste võidakse soovitada veresugulastele. George Huntington kirjeldas seda seisundit esmakordselt 1872. aastal pärast Long Islandi elanike perekondliku juhtumi uurimist.
Huntingtoni tõve levimus on ligikaudu 10 juhtu 100 000 elaniku kohta ja arvestades selle hilist algust, on ligikaudu 30 inimesel 100 000-st 50% risk selle elu jooksul haigestuda. Kuigi haigus ilmneb kõige sagedamini 35–40-aastaselt, on haigestumise vanusevahemik üsna lai, kusjuures varaseim algus on 3-aastaselt ja hiliseim 90-aastaselt. Kuigi algselt arvati, et haiguse penetrantsus on 100%, arvatakse nüüd, et see pole alati nii. Isikutel, kes pärisid haiguse geeni oma isalt, avaldub haigus keskmiselt 3 aastat varem kui neil, kes pärisid patoloogilise geeni oma emalt. Ligikaudu 80%-l patsientidest, kes pärisid patoloogilise geeni oma isalt, avaldub haigus enne 20. eluaastat. Geneetilise defekti varasema avaldumise nähtust järglastel nimetatakse ennetamiseks.
 [ 1 ]
[ 1 ]
Mis põhjustab Huntingtoni tõbe?
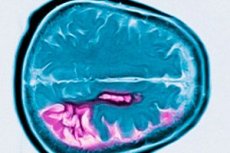
Huntingtoni tõvel puudub sooline eelistus. Näidatakse sabatuuma atroofiat, kus väikesed neuronid degenereeruvad ja neurotransmitterite - gamma-aminovõihappe (GABA) ja substantsi P - tase langeb.
Huntingtoni tõve tekke eest vastutab mutantgeen, millel on suurenenud arv ("laienemine") CAG (tsüsteiin-alaniin-glütsiin) DNA järjestusi, mis kodeerivad aminohapet glutamiini. Selle geeni produkt, suur valk huntingtin, sisaldab liigselt polüglutamiinijääke, mis viib haiguseni tundmatu mehhanismi kaudu. Mida rohkem CAG kordusi, seda varem haigus avaldub ja seda raskem on selle kulg. Põlvkonnast põlve võib korduste arv suureneda, mis aja jooksul viib perekonna fenotüübi süvenemiseni.
Vaatamata märkimisväärsele huvile Parkinsoni tõve geneetiliste ja biokeemiliste muutuste vastu, ei õnnestunud haiguse geeni otsingud 1970. aastate lõpuni. Sel ajal korraldasid Nancy Wexler ja Allan Tobin Pärilike Haiguste Fondi sponsoreeritud seminari, et arutada Huntingtoni tõve geeni leidmise strateegiat. Koosolekul osalenud David Housman, David Botstein ja Ray White pakkusid välja, et hiljuti väljatöötatud rekombinantse DNA meetodid võivad aidata seda eesmärki saavutada. Projekti põhiülesanne oli leida suur perekond, kus on palju Huntingtoni tõve põlvkondi, et saada DNA proove. 1979. aastal käivitati Venezuela ja Ameerika Ühendriikide teadlaste ühisprojekt, et uurida suurt Huntingtoni tõvega perekonda, kes elab Maracheibo järve kaldal (Venezuela). 1983. aastal lokaliseeriti Huntingtoni tõve geen 4. kromosoomi lühikese haru lõpus (Gusella jt, 1983) ja kümme aastat hiljem selgus, et selle geeni mutatsioon seisneb tsütosiin-adeniin-guaniini (CAG) trinukleotiidi korduste arvu suurenemises (Huntingtoni tõve koostöörühm, 1993). Selle teadusrühma väljatöötatud metoodikat peetakse praegu uute geenide positsioonilise kloonimise standardiks.
Kuigi metsiktüüpi geenil on 10–28 CAG-korduse pikkune piirkond, on Huntingtoni tõbe põhjustava geeni mutantsel vormil CAG-korduste arv suurenenud 39-lt enam kui 100-le. Trinukleotiidide korduste laienemise avastamine on aidanud selgitada paljusid haiguse kliinilisi tunnuseid. Eelkõige leiti pöördvõrdeline seos haiguse alguse vanuse ja korduvate trinukleotiididega piirkonna pikkuse vahel. Isapoolse pärandumise eeldamist saab seletada asjaoluga, et meestel toimub spermatogeneesi ajal sageli korduste arvu suurenemine. Uute mutatsioonide analüüs on näidanud, et need tekivad tavaliselt siis, kui ühel vanematest, tavaliselt isal, oli CAG-korduste arv suurem kui 28; sel juhul suurenes nende korduste arv järgmises põlvkonnas. Nüüdseks on kindlaks tehtud, et kui korduste arv ei ole suurem kui 28, kandub see stabiilselt põlvest põlve. Kui korduste arv on 29 kuni 35, siis Huntingtoni tõve sümptomeid ei avaldu, kuid järglastele edasikandumisel võib selle piirkonna pikkus suureneda. Kui korduste arv on 36 kuni 39, siis mõnel juhul (kuid mitte alati) võib haigus avalduda kliiniliselt (mittetäielik penetrance) ja järglastele edasikandumisel on võimalik trinukleotiidide korduste arvu suurenemine. Kui korduste arv ületab 40, siis esineb haigus peaaegu kõigil juhtudel ja järglastele edasikandumisel on võimalik korduste edasine laienemine. Korduste arvu suurenemise põhjused on teadmata.
Huntingtoni tõve patomorfoloogia
Huntingtoni tõbe iseloomustab neuronite kadu peamiselt sabatuumas ja putamenis ning teatud määral ka ajukoores ja teistes ajustruktuurides. Huntingtoni tõve korral väheneb aju kogukaal mitte ainult neuronite arvu vähenemise, vaid ka valgeaine kao tõttu. Ajukoores on enim mõjutatud V ja VI kihi rakud. Mikro- ja makroskoopiliste degeneratiivsete muutuste raskusaste (kohandatud surmaeaga) korreleerub CAG korduste arvuga. Mitmesaja Huntingtoni tõve juhtumi muutuste detailne patoloogiline analüüs on näidanud, et striatumi degeneratsioon algab sabatuumas dorsomediaalses osas ja putamenis dorsolateraalses osas ning levib seejärel ventraalselt. Erinevad neuronite rühmad sabatuumas ja putamenis on mõjutatud erineval määral. Striatumi interneuronid jäävad suhteliselt terveks, kuid mõned projektsioonneuronid on selektiivselt mõjutatud. Huntingtoni tõve juveniilse vormi korral on striatumis esinevad patomorfoloogilised muutused selgemalt väljendunud ja laialdasemad, hõlmates ajukoore, väikeaju, talamuse ja globus pallidus'e.
Huntingtoni tõve neurokeemilised muutused
GABA. Huntingtoni tõvega patsientide aju neurokeemilised uuringud näitasid GABA kontsentratsiooni olulist langust striatumis. Hilisemad uuringud kinnitasid, et Huntingtoni tõbi on seotud GABAergiliste neuronite arvu vähenemisega ja näitasid, et GABA kontsentratsioon väheneb mitte ainult striatumis, vaid ka selle projektsioonitsoonides - globus pallidus'e välis- ja sisesegmentides ning substantia nigras. Huntingtoni tõvega ajus tuvastati GABA retseptorite muutusi ka retseptorite sidumise uuringute ja mRNA in situ hübridisatsiooni abil. GABA retseptorite arv oli mõõdukalt vähenenud sabatuumas ja putamenis, kuid suurenenud substantia nigra retikulaarses osas ja globus pallidus'e välissegmendis, mis on tõenäoliselt tingitud denervatsiooni ülitundlikkusest.
Atsetüülkoliin. Atsetüülkoliini kasutavad neurotransmitterina suured mitte-okkalised interneuronid striatumis. Varased surmajärgsed uuringud Huntingtoni tõvega patsientidel näitasid koliini atsetüültransferaasi (ChAT) aktiivsuse vähenemist striatumis, mis viitab kolinergiliste neuronite kadumisele. Võrreldes GABAergiliste neuronite olulise vähenemisega on kolinergilised interneuronid aga suhteliselt säästetud. Seetõttu on atsetüülkoliinesteraas-positiivsete neuronite tihedus ja ChAT aktiivsus striatumis tegelikult suhteliselt kõrgenenud võrreldes samaealiste kontrollrühmadega.
Aine P. Aine P-d leidub paljudes striatumi keskmise pikkusega okastega neuronites, mis ulatuvad peamiselt globus pallidus'e sisemisse segmenti ja mustaineossa ning sisaldavad tavaliselt ka dünorfiini ja GABA-d. Huntingtoni tõve korral on substantia P tase striatumis ja mustaineosa pars reticularis'es vähenenud. Haiguse lõppstaadiumis on immunohistokeemilised uuringud näidanud substantia P-d sisaldavate neuronite arvu olulist vähenemist. Varasemates staadiumides on substantia P-d sisaldavad ja globus pallidus'e sisemisse segmenti ulatuvad neuronid suhteliselt säästetud võrreldes neuronitega, mis ulatuvad mustaineosa pars reticularis'esse.
Opioidpeptiidid. Enkefaliin sisaldub kaudse raja keskmise okastega projektsiooniga GABAergilistes neuronites, mis ulatuvad globus pallidus'e välissegmenti ja kannavad D2-retseptoreid. Immunohistokeemilised uuringud on näidanud, et globus pallidus'e välissegmenti ulatuvad enkefaliini sisaldavad neuronid kaovad Huntingtoni tõve alguses. Need rakud surevad ilmselt varem kui substants P-d sisaldavad rakud, mis ulatuvad globus pallidus'e sisesegmenti.
Katehhoolamiinid. Biogeenseid amiine (dopamiin, serotoniin) sisaldavad ja striatumisse ulatuvad neuronid paiknevad mustaine kompaktses osas, ventraalses tegmentumis ja raphe tuumades. Kuigi noradrenergilised projektsioonid inimese striatumisse on minimaalsed, on serotoniini ja dopamiini tase (grammi koe kohta) striatumi sees kõrgenenud, mis näitab nende aferentsete projektsioonide säilimist vaatamata striatumi enda neuronite märkimisväärsele kadumisele. Mustaine dopamiinergilised neuronid jäävad terveks nii Huntingtoni tõve klassikalises kui ka juveniilses vormis.
Somatostatiin/neuropeptiid Y ja lämmastikoksiidi süntetaas. Huntingtoni tõvega patsientidel striatumis mõõdetud somatostatiini ja neuropeptiid Y tasemed näitasid 4-5-kordset suurenemist võrreldes normaalsete kudedega. Immunohistokeemilised uuringud näitasid neuropeptiid Y-d, somatostatiini ja lämmastikoksiidi süntetaasi sisaldavate striataalsete interneuronite absoluutset säilimist. Seega on need neuronid patoloogilise protsessi suhtes resistentsed.
Ergutavad aminohapped. On oletatud, et Huntingtoni tõve selektiivne rakusurm on tingitud glutamaadi poolt indutseeritud neurotoksilisest toimest. Glutamaadi ja kinoliinhappe (endogeenne neurotoksiin, mis on serotoniini metabolismi kõrvalprodukt ja glutamaadi retseptorite agonist) tasemed Huntingtoni tõve striatumis on veidi muutunud, kuid hiljutine MR-spektroskoopia abil läbi viidud uuring näitas glutamaadi taseme tõusu in vivo. Kinoliinhappe sünteesi eest vastutava gliaalensüümi tase Huntingtoni tõve striatumis on normaalsega võrreldes umbes 5 korda suurenenud, samas kui kinoliinhappe lagundamist tagava ensüümi aktiivsus on Huntingtoni tõve korral suurenenud vaid 20–50%. Seega võib kinoliinhappe süntees Huntingtoni tõve korral olla suurenenud.
Ergutavate aminohapete (EAA) retseptorite uuringud Huntingtoni tõve korral on näidanud NMDA, AMPA, kainaadi ja metabotroopsete glutamaadi retseptorite arvu olulist vähenemist striatumis, samuti AMPA ja kainaadi retseptorite arvu olulist vähenemist ajukoores. Huntingtoni tõve hilisemas staadiumis NMDA retseptorid praktiliselt puudusid, samas kui prekliinilises ja varases staadiumis täheldati nende retseptorite arvu olulist vähenemist.
Selektiivne tundlikkus. Huntingtoni tõve korral kaovad teatud tüüpi striataalsed rakud selektiivselt. Keskmise suurusega okastega neuronid, mis ulatuvad globus pallidus'e välissegmenti ja sisaldavad GABA-d ja enkefaliini, surevad haiguse väga varakult, nagu ka GABA-d ja substantsi P sisaldavad neuronid, mis ulatuvad mustaine retikulaarsesse ossa. GABA-d ja enkefaliini sisaldavate ning globus pallidus'e välissegmenti ulatuvate neuronite kadumine disinhibeerib seda struktuuri, mis omakorda viib subtalamuse tuuma aktiivse pärssimiseni. Subtalamuse tuuma vähenenud aktiivsus võib ilmselt selgitada Huntingtoni tõve korral esinevaid koreaformseid liigutusi. On juba ammu teada, et subtalamuse tuuma fokaalsed kahjustused võivad põhjustada koread. GABA ja substantia nigra pars reticularis'esse ulatuvate neuronite kadumine on tõenäoliselt vastutav Huntingtoni tõve korral täheldatud silmamotoorika häirete eest. See rada pärsib tavaliselt mustainetüki pars retikulaarse neuroneid, mis ulatuvad ülemisse kollikulusse ja mis omakorda reguleerivad sakkaade. Juveniilse Huntingtoni tõve korral on eespool mainitud rajad tugevamalt mõjutatud ja lisaks kaovad varakult striatumi projektsioonid globus pallidus'e sisemisele segmendile.
Huntingtoni tõbe põhjustava geeni poolt kodeeritud valk huntingtin esineb aju ja teiste kudede erinevates struktuurides. Tavaliselt leidub huntingtiini peamiselt neuronite tsütoplasmas. Valku leidub enamikus aju neuronites, kuid hiljutised andmed näitavad, et selle sisaldus on maatriksneuronites suurem kui striosoomi neuronites ja projektsioonneuronites suurem kui interneuronites. Seega korreleerub neuronite selektiivne tundlikkus nende huntingtiini sisaldusega, mis on tavaliselt teatud neuronite populatsioonides olemas.
Nagu Huntingtoni tõvega patsientide ajudes, moodustab huntingtin ka hiirtel, kes on transgeensed Huntingtoni tõve geeni N-terminaalse fragmendi suhtes, millel on laiendatud korduste arv, neuronite tuumades tihedaid agregaate. Need tuumasisesed inklusioonid moodustuvad striatumi projektsioonneuronites (kuid mitte interneuronites). Transgeensetel hiirtel moodustuvad inklusioonid mitu nädalat enne sümptomite ilmnemist. Need andmed viitavad sellele, et suurenenud arvu glutamiinijääke sisaldav huntingtinvalk, mille inklusioonid kodeerivad trinukleotiidide kordusi või selle fragmenti, akumuleerub tuumas ja võib seega kahjustada selle kontrolli rakuliste funktsioonide üle.
Huntingtoni tõve sümptomid
Huntingtoni tõvega patsientidel on esimeste sümptomite ilmnemise vanust raske täpselt kindlaks määrata, kuna haigus avaldub järk-järgult. Isiksuse ja käitumise muutused, kerged koordinatsioonihäired võivad ilmneda aastaid enne selgemate sümptomite ilmnemist. Diagnoosi panemise ajaks on enamikul patsientidest koreaalsed liigutused, peenliigutuste koordinatsioonihäired ja tahtele alluvate sakaadide aeglane genereerimine. Haiguse progresseerumisel halveneb võime oma tegevusi korraldada, mälu väheneb, kõne muutub raskeks, silmamotoorika häired ja koordineeritud liigutuste soorituse häired suurenevad. Kuigi haiguse algstaadiumis lihastes ja kehahoiakus muutusi ei toimu, võivad progresseerudes tekkida düstoonilised kehahoiakud, mis aja jooksul võivad muutuda domineerivaks sümptomiks. Hilises staadiumis muutub kõne ebaselgeks, neelamine muutub oluliselt raskemaks, kõndimine muutub võimatuks. Huntingtoni tõbi progresseerub tavaliselt 15-20 aasta jooksul. Terminaalses staadiumis on patsient abitu ja vajab pidevat hooldust. Surmaga lõppev tulemus ei ole otseselt seotud põhihaigusega, vaid selle tüsistustega, näiteks kopsupõletikuga.
Huntingtoni tõve dementsus
RHK-10 kood
P02.2. Huntingtoni tõvega seotud dementsus (G10).
Dementsus areneb süsteemse degeneratiivse-atroofilise protsessi ühe ilminguna, mille puhul on valdavalt kahjustatud aju striataalne süsteem ja teised subkoekaalsed tuumad. See pärandub autosomaalselt dominantsel viisil.
Reeglina avaldub haigus elu kolmandal või neljandal kümnendil koreoformse hüperkineesiga (eriti näos, kätes, õlgades, kõnnakus), isiksuse muutustega (erutunud, hüsteerilised ja skisoidsed isiksuseomaaliad), psühhootiliste häiretega (eriline depressioon koos sünguse, pahuruse, düsfooriaga; paranoiline meeleolu).
Diagnostika seisukohalt on eriti oluline koreoformse hüperkineesi, dementsuse ja päriliku koormuse kombinatsioon. Järgnev on spetsiifiline sellele dementsusele:
- aeglane progresseerumine (keskmiselt 10–15 aastat): dissotsiatsioon säilinud enesehooldusvõime ja ilmse intellektuaalse ebakompetentsuse vahel olukordades, mis nõuavad produktiivset vaimset tööd (kontseptuaalne mõtlemine, uute asjade õppimine);
- vaimse soorituse väljendunud ebaühtlus, mis põhineb tähelepanu jämedatel häiretel ja patsiendi hoiakute ebajärjekindlusel ("tõmblev" mõtlemine, sarnane hüperkineesiga);
- kõrgemate kortikaalsete funktsioonide ilmsete rikkumiste atüüpilisus;
- dementsuse sagenemise ja psühhootiliste häirete raskusastme vahel on pöördvõrdeline seos.
Võttes arvesse psühhootiliste (paranoilised armukadeduse, tagakiusamise luulud) ja düsfooriliste häirete suurt osakaalu haiguse kliinilises pildis, viiakse ravi läbi erinevate neuroleptikumide abil, mis blokeerivad dopamiinergilisi retseptoreid (fenotiasiini ja butürofenooni derivaadid) või vähendavad dopamiini taset kudedes (reserpiin).
Kasutatakse haloperidooli (2–20 mg/päevas), tiapriidi (100–600 mg/päevas) mitte kauem kui kolm kuud, tioridasiini (kuni 100 mg/päevas), reserpiini (0,25–2 mg/päevas) ja krambivastast ravimit klonasepaami (1,5–6 mg/päevas). Need ravimid aitavad vähendada hüperkineesi, siluda afektiivset pinget ja kompenseerida isiksusehäireid.
Vaimse häire statsionaarne ravi viiakse läbi, võttes arvesse patsiendi juhtivat sündroomi, vanust ja üldist seisundit. Ambulatoorses ravis on teraapia põhimõtted samad (liikumishäirete pidev säilitusravi, ravimi perioodiline vahetamine). Ambulatoorses ravis kasutatakse neuroleptikumide väiksemaid annuseid.
Kerge ja mõõduka dementsuse rehabilitatsioonimeetmete hulka kuuluvad ergoteraapia, psühhoteraapia ja kognitiivne treening. Vajalik on koostöö pereliikmetega ja patsiendi eest hoolitsevate inimeste psühholoogilise toe pakkumine. Haiguse ennetamise peamine meetod on lapse saamise otsustamisel patsiendi lähimate sugulaste meditsiiniline ja geneetiline nõustamine koos saatekirjaga DNA-analüüsiks.
Prognoos on üldiselt ebasoodne. Haiguse kulg on aeglaselt progresseeruv ja haigus viib tavaliselt surmani 10-15 aasta pärast.
[ 18 ]
Mis teid häirib?
Huntingtoni tõve ravi
Huntingtoni tõve ravi on sümptomaatiline. Koread ja agitatsiooni saab osaliselt pärssida neuroleptikumidega (nt klorpromasiin 25–300 mg suu kaudu 3 korda päevas, haloperidool 5–45 mg suu kaudu 2 korda päevas) või reserpiiniga 0,1 mg suu kaudu üks kord päevas. Annuseid suurendatakse talutava maksimumini (enne kõrvaltoimete, näiteks unisuse, parkinsonismi; reserpiini puhul hüpotensiooni, tekkimist). Empiirilise ravi eesmärk on vähendada glutamaatergilist ülekannet Nmetüül-O-aspartaadi retseptorite kaudu ja säilitada energiatootmine mitokondrites. Ravi, mille eesmärk on suurendada GABA taset ajus, on ebaefektiivne.
Geneetiline testimine ja nõustamine on olulised, kuna haiguse sümptomid ilmnevad pärast sünnitusiga. Positiivse perekonnaanamneesiga inimesed ja testimisest huvitatud inimesed suunatakse spetsialiseeritud keskustesse, võttes arvesse kõiki eetilisi ja psühholoogilisi aspekte.
Huntingtoni tõve sümptomaatiline ravi
Huntingtoni tõve progresseerumist peatav efektiivne ravi puudub. Erinevate ravimitega on läbi viidud mitmeid uuringuid, kuid märkimisväärset efekti pole saavutatud. Neuroleptikume ja teisi dopamiini retseptori antagoniste kasutatakse laialdaselt vaimsete häirete ja tahtmatute liigutuste korrigeerimiseks Huntingtoni tõvega patsientidel. Tahtmatud liigutused peegeldavad dopamiinergilise ja GABAergilise süsteemi tasakaalustamatust. Seetõttu kasutatakse neuroleptikume liigse dopamiinergilise aktiivsuse vähendamiseks. Need ravimid ise võivad aga põhjustada olulisi kognitiivseid ja ekstrapüramidaalseid kõrvaltoimeid. Lisaks, välja arvatud juhtudel, kui patsiendil tekib psühhoos või agitatsioon, pole nende efektiivsust tõestatud. Neuroleptikumid põhjustavad või süvendavad sageli düsfaagiat või muid liikumishäireid. Uuema põlvkonna neuroleptikumid, nagu risperidoon, kloasapiin ja olansapiin, võivad olla Huntingtoni tõve ravis eriti kasulikud, kuna need põhjustavad vähem ekstrapüramidaalseid kõrvaltoimeid, kuid võivad vähendada paranoilisi sümptomeid või suurenenud ärrituvust.
Tetrabenasiin ja reserpiin vähendavad samuti dopamiinergilise süsteemi aktiivsust ja võivad haiguse algstaadiumis vähendada tahtmatute liigutuste raskust. Siiski võivad need ravimid põhjustada depressiooni. Kuna haigus ise põhjustab sageli depressiooni, piirab see kõrvaltoime oluliselt reserpiini ja tetrabenasiini kasutamist. Haiguse hilisemas staadiumis dopamiiniretseptoreid kandvad rakud surevad, mistõttu dopamiiniretseptori antagonistide efektiivsus nõrgeneb või kaob.
Huntingtoni tõvega patsientidel kasutatakse psühhoosi, depressiooni ja ärrituvuse raviks neuroleptikume, antidepressante ja anksiolüütikume, kuid neid tuleks määrata ainult seni, kuni patsiendil need sümptomid tegelikult esinevad. Ravimid, mis võivad haiguse ühes staadiumis abiks olla, võivad haiguse progresseerudes muutuda ebaefektiivseks või isegi kahjulikuks.
GABA retseptori agoniste on testitud Huntingtoni tõvega patsientidel, kuna Huntingtoni tõve puhul on näidatud GABA taseme olulist langust striatumis, samuti GABA retseptorite ülitundlikkust selle projektsioonipiirkondades. Bensodiasepiinid on osutunud tõhusaks juhtudel, kus stressi ja ärevuse tõttu süvenevad tahtmatud liigutused ja kognitiivsed häired. Nende ravimite väikeseid annuseid tuleks määrata soovimatute rahustavate toimete vältimiseks. Enamikul Huntingtoni tõvega patsientidest ei põhjusta ükski ravim elukvaliteedi olulist paranemist.
Parkinsoni tõve sümptomitega Huntingtoni tõve varajases staadiumis võib proovida dopamiinergilisi aineid, kuid nende efektiivsus on piiratud. Lisaks võib levodopa nendel patsientidel põhjustada või süvendada müokloonust. Samal ajal võib baklofeen mõnedel Huntingtoni tõvega patsientidel vähendada lihasjäikust.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Huntingtoni tõve ennetav (neuroprotektiivne) ravi
Kuigi Huntingtoni tõve geneetiline defekt on teada, on endiselt ebaselge, kuidas see viib selektiivse neuronaalse degeneratsioonini. Hüpotees on, et oksüdatiivse stressi ja eksitotoksilisuse vähendamisele suunatud ennetavad ravimeetodid võivad potentsiaalselt haiguse progresseerumist aeglustada või peatada. Olukord võib olla mõnevõrra sarnane hepatolentikulaarse degeneratsiooniga, mille puhul geneetiline defekt jäi aastaid tundmatuks, kuid sekundaarsele toimele, vase akumuleerumisele, suunatud ennetavad ravimeetodid "raviti". Sellega seoses on erilist tähelepanu pälvinud hüpotees, et Huntingtoni tõbi on seotud energia metabolismi häire ja eksitotoksilisusest tingitud rakusurmaga. Haigus ise võib põhjustada rakusurma huntingtiini N-terminaalsete fragmentide intranukleaarse agregatsiooni tõttu, mis häirib rakkude ja ainevahetuse funktsioone. See protsess võib mõjutada mõningaid neuronirühmi suuremal määral kui teisi, kuna need on eksitotoksilise kahjustuse suhtes tundlikumad. Sellisel juhul suudab ennetav ravi ergastavate aminohappe retseptori antagonistide või vabade radikaalide kahjustusi ennetavate ainetega haiguse algust ja progresseerumist ennetada või edasi lükata. Amüotroofse lateraalskleroosi laboratoorsetes mudelites on näidatud, et antioksüdandid ja retseptori antagonistid (RAA-d) suudavad haiguse progresseerumist aeglustada. Sarnased lähenemisviisid võivad olla tõhusad Huntingtoni tõve puhul. Praegu on käimas kliinilised uuringud glutamaadi retseptori antagonistide ja mitokondriaalse elektronide transpordiahela II kompleksi funktsiooni võimendavate ainete kohta.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]