Artikli meditsiiniline ekspert

Uued väljaanded
Usheri sündroom
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
Usheri sündroom on pärilik haigus, mis avaldub täieliku sünnist saati tekkinud kurtusena ja vanusega progresseeruva pimedusena. Nägemise kaotus on seotud pigmentretiniidiga, mis on võrkkesta pigmendi degeneratsiooni protsess. Paljudel Usheri sündroomiga inimestel on ka rasked tasakaaluprobleemid.
Epidemioloogia
Tänu uuringule oli võimalik kindlaks teha, et Usheri sündroom mõjutab umbes 8% uuritud kurttummadest lastest (testid viidi läbi kurttummadele mõeldud spetsiaalsetes asutustes). Pigmentaarset retiniiti täheldati 6–10%-l kaasasündinud kurtusega patsientidest, mis omakorda esineb umbes 30%-l pigmentaarse võrkkesta haigusega inimestest.
Arvatakse, et see haigus avaldub ligikaudu 3–10 inimesel 100 tuhandest kogu maailmas. Seda võib täheldada võrdselt nii naistel kui ka meestel. Selle sündroomi all kannatab umbes 5–6% maailma elanikkonnast. Umbes 10% kõigist lapsepõlves esineva sügava kurtuse juhtudest on tingitud nii Usheri sündroomi I kui ka II tüübist.
Ameerika Ühendriikides on 1. ja 2. tüüp kõige levinumad. Koos moodustavad need ligikaudu 90–95 protsenti kõigist Usheri sündroomi juhtudest lastel.
Põhjused Usheri sündroom
Usheri sündroomi tüübid I, II ja III on autosoom-retsessiivselt päranduvad, samas kui IV tüüpi peetakse X-kromosoomi häireks. Selle sündroomiga kaasneva pimeduse ja kurtuse põhjuseid ei ole veel piisavalt uuritud. Eeldatakse, et selle haigusega inimesed on ülitundlikud komponentide suhtes, mis võivad kahjustada DNA struktuuri. Lisaks võib see haigus olla seotud immuunsüsteemi häiretega, kuid antud juhul puudub selle protsessi täpne pilt.
1989. aastal tuvastati II tüüpi haigusega patsientidel esmakordselt kromosoomanomaaliad, mis võib tulevikus viia sündroomi põhjustavate geenide isoleerimiseni. Samuti võib olla võimalik neid geene kandjatel tuvastada ja spetsiaalseid sünnieelseid geneetilisi teste välja töötada.
 [ 8 ]
[ 8 ]
Riskitegurid
Sündroom pärandub, kui mõlemad vanemad on haigestunud, st see pärandub retsessiivselt. Laps võib haiguse pärida ka siis, kui tema vanemad on geeni kandjad. Kui mõlemal tulevasel vanemal on see geen, on selle sündroomiga lapse saamise tõenäosus 1:4. Inimest, kellel on ainult üks sündroomi geen, peetakse kandjaks, kuid tal ei esine häire sümptomeid. Tänapäeval ei ole veel võimalik kindlaks teha, kas inimesel on selle haiguse geen olemas.
Kui laps sünnib vanematele, kellest ühel sellist geeni pole, on sündroomi pärimise tõenäosus väga väike, kuid ta on kindlasti kandja.
Sümptomid Usheri sündroom
Usheri sündroomi sümptomiteks on kuulmislangus ja pigmenteerunud rakkude ebanormaalne kogunemine silma struktuuridesse. Seejärel tekib patsiendil võrkkesta degeneratsioon, mis põhjustab nägemise halvenemist ja kõige raskematel juhtudel nägemise kaotust.
Sensorineuraalne kuulmislangus võib olla kerge või täielik ja tavaliselt ei progresseeru sünnist saati. Võrkkesta pigmendihaigus võib aga hakata tekkima juba lapsepõlves või hiljem. Testide tulemused on näidanud, et tsentraalne nägemisteravus võib säilida aastaid isegi siis, kui perifeerne nägemine halveneb (seisund, mida nimetatakse tunnelinägemiseks).
Need on haiguse peamised ilmingud, millele võivad mõnikord lisanduda muud häired, näiteks psühhoos ja muud vaimsed häired, sisekõrva probleemid ja/või katarakt.
Vormid
Uuringu käigus tuvastati selle haiguse 3 tüüpi, samuti neljas vorm, mis on üsna haruldane.
Haiguse I tüüpi iseloomustab kaasasündinud täielik kurtus ja tasakaaluhäired. Sageli hakkavad sellised lapsed kõndima alles 1,5-aastaselt. Nägemise halvenemine algab tavaliselt 10-aastaselt ja ööpimeduse lõplik areng algab 20-aastaselt. Seda tüüpi haigusega lastel võib tekkida perifeerse nägemise progresseeruv halvenemine.
II tüüpi haiguse korral täheldatakse mõõdukat või kaasasündinud kurtust. Sellisel juhul osalise kurtuse halvenemist enam sageli ei esine. Pigmentaarne retiniit hakkab tekkima noorukiea lõpus või 20 aasta pärast. Ööpimeduse teke algab tavaliselt 29–31 aasta vanuselt. Nägemisteravuse halvenemine II tüüpi patoloogia korral progresseerub üldiselt veidi aeglasemalt kui I tüübi korral.
Haiguse III tüüpi iseloomustab progresseeruv kuulmislangus, mis algab tavaliselt puberteedieas, ning samal perioodil (veidi hiljem kui kuulmislangus) järkjärguline retiniidi pigmentosa teke, mis võib muutuda progresseeruva pimeduse tekke teguriks.
IV tüüpi patoloogia ilmingud esinevad peamiselt meestel. Sellisel juhul täheldatakse ka progresseeruvaid häireid ning kuulmis- ja nägemiskaotust. See vorm on väga haruldane ja tavaliselt X-kromosomaalse iseloomuga.
Diagnostika Usheri sündroom
Usheri sündroomi diagnoos pannakse patsiendi täheldatud äkilise kurtuse ja progresseeruva nägemiskaotuse kombinatsiooni põhjal.
Testid
Mutatsiooni tuvastamiseks võidakse tellida spetsiaalne geneetiline test.
On leitud üksteist geneetilist lookust, mis võivad põhjustada Usheri sündroomi teket, ja on tuvastatud üheksa geeni, mis on kindlasti häire põhjuseks:
- Tüüp 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tüüp 2: ush2a, VLGR1, WHRN.
- Usheri sündroomi tüüp 3: USH3A.
NIDCD teadlased koos New Yorgi ja Iisraeli ülikoolide kolleegidega on tuvastanud Pcdh15 geenis mutatsiooni nimega R245X, mis põhjustab juudi populatsioonis suure osa 1. tüüpi Usheri sündroomist.
Kliinilisi uuringuid läbiviivate laborite kohta lisateabe saamiseks külastage veebisaiti https://www.genetests.org ja otsige laborikataloogist märksõna "Usheri sündroom".
Usheri sündroomi geneetilist testimist hõlmavate olemasolevate kliiniliste uuringute kohta lisateabe saamiseks külastage veebisaiti https://www.clinicaltrials.gov ja otsige „Usheri sündroom” või „Usheri sündroomi geneetiline testimine”.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentaalne diagnostika
Instrumentaalse diagnostika jaoks on mitu meetodit:
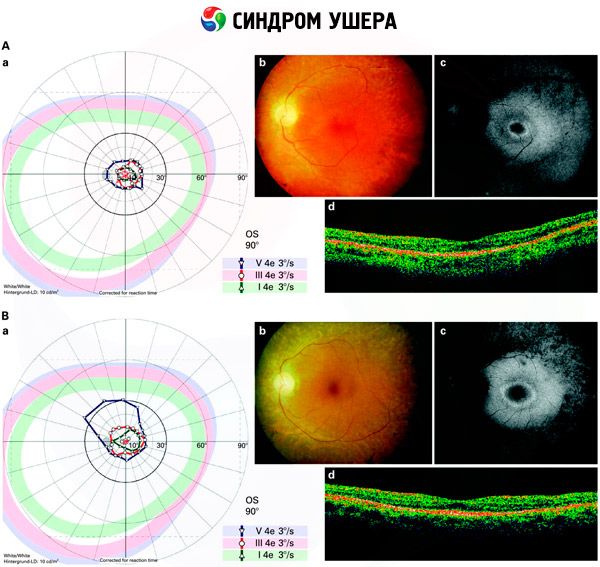
- Silmapõhja uurimine võrkkesta pigmendilaikude olemasolu tuvastamiseks, samuti võrkkesta veresoonte ahenemine;
- Elektroretinogramm, mis võimaldab tuvastada silma võrkkesta esialgseid degeneratiivseid kõrvalekaldeid. See näitab elektroradiograafiliste radade hääbumist;
- Elektronüstagmogramm (ENG) mõõdab tahtmatuid silmaliigutusi, mis võivad viidata tasakaalutuse olemasolule.
- Audiomeetria, mida kasutatakse kurtuse olemasolu ja selle raskusastme kindlakstegemiseks.
Diferentseeritud diagnoos
Usheri sündroomi tuleb eristada mõnest sarnasest häirest.
Hallgreni sündroom, mida iseloomustab kaasasündinud kuulmislangus ja progresseeruv nägemiskaotus (tekivad ka katarakt ja nüstagm). Lisasümptomite hulka kuuluvad ataksia, psühhomotoorsed häired, psühhoos ja vaimne alaareng.
Alströmi sündroom, mis on pärilik haigus, mille korral võrkkest degenereerub, mille tulemuseks on keskse nägemise kaotus. See sündroom on seotud laste rasvumisega. Samal ajal hakkavad 10 aasta pärast tekkima suhkurtõbi ja kuulmislangus.
Punetised rasedal naisel esimesel trimestril võivad põhjustada lapse arengus mitmesuguseid kõrvalekaldeid. Sellise kõrvalekalde tagajärgede hulka kuuluvad kuulmislangus, samuti (või) nägemisprobleemid ja lisaks sellele mitmesugused arenguvead.
Kellega ühendust võtta?
Ravi Usheri sündroom
Usheri sündroomile ei ole praegu ravi. Seetõttu seisneb ravi sel juhul peamiselt nägemiskaotuse protsessi aeglustamises ja kuulmislanguse kompenseerimises. Võimalike ravimeetodite hulka kuuluvad:
- A-vitamiini võtmine (mõned silmaarstid usuvad, et A-vitamiini palmitaadi suured annused võivad aeglustada, kuid mitte peatada pigmentretiniidi progresseerumist);
- Spetsiaalsete elektrooniliste seadmete (kuuldeaparaadid, kohleaarimplantaadid) paigaldamine patsiendi kõrva.
Silmaarstid soovitavad enamikul täiskasvanutel, kellel esineb pigmentretiniidi tavalisi vorme, võtta iga päev 15 000 RÜ (rahvusvahelist ühikut) A-vitamiini palmitaati järelevalve all. Kuna uuringusse ei kaasatud 1. tüüpi Usheri sündroomiga inimesi, ei ole sellele patsientide rühmale soovitatav A-vitamiini suuri annuseid. Inimesed, kes kaaluvad A-vitamiini võtmist, peaksid seda ravivõimalust oma arstiga arutama. Muud soovitused selle ravivõimaluse kohta on järgmised:
- Muutke oma dieeti, et see sisaldaks A-vitamiinirikkaid toite.
- Rasedaks jääda plaanivad naised peaksid A-vitamiini suurte annuste võtmise lõpetama kolm kuud enne planeeritud rasestumist, kuna see suurendab sünnidefektide riski.
- Rasedad naised peaksid A-vitamiini suurte annuste võtmise lõpetama suurenenud sünnidefektide riski tõttu.
Samuti on oluline sellist last sotsiaalse eluga kohaneda. Selleks on vaja eripedagoogide ja psühholoogide abi. Juhul, kui patsiendil on hakanud tekkima järkjärguline nägemiskaotus, tuleks talle õpetada viipekeelt kasutama.
Prognoos
Usheri sündroomil on ebasoodne prognoos. Nägemisväli ja selle teravus hakkavad enamikul selle haigusega patsientidest halvenema 20–30 aasta jooksul. Mõnel juhul tekib täielik kahepoolne nägemiskaotus. Kuulmislangus, millega alati kaasneb tummus, areneb väga kiiresti täielikuks kahepoolseks kuulmislanguseks.