Artikli meditsiiniline ekspert

Uued väljaanded
Pierre Robini sündroom
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.

Pierre-Robini sündroom, meditsiinis tuntud ka kui Robini anomaalia, on näo lõualuu arengu kaasasündinud patoloogia. Haigus sai oma nime prantsuse hambaarsti P. Robini auks, kes kirjeldas esimesena kõiki selle sümptomeid. Lannelongue ja Menard kirjeldasid Pierre-Robini sündroomi esmakordselt 1891. aastal oma aruandes kahe mikrognaatia, suulaelõhe ja retroglossoptoosiga patsiendi kohta. 1926. aastal avaldas Pierre-Robin haiguse juhtumi imikul, kellel esinesid klassikalise sündroomi tunnused. Kuni 1974. aastani oli see tunnuste triaad tuntud kui Robin-Pierre'i sündroom. Nüüd aga kasutatakse seda sündroomi mitmete anomaaliate samaaegse esinemisega väärarengute kirjeldamiseks.
Epidemioloogia
See on heterogeenne kaasasündinud defekt, mille esinemissagedus on 1 8500 elussünni kohta. Meeste ja naiste suhe on 1:1, välja arvatud X-kromosoomiga seotud vorm.
Nende patsientide seas on 50%-l imikutest mittetäielik pehmesuulaelõhe, ülejäänud sünnivad kaarja ja ebatavaliselt kõrge suulaelõhega, kuid ilma lõheta.
Põhjused Pierre Robini sündroom
Arvesse võetakse haiguse autosomaalse retsessiivse pärandumise võimalust. Sõltuvalt etioloogiast on kahte tüüpi sündroomi: isoleeritud ja geneetiliselt määratud. Isoleeritud tüüp tekib lõualuu alaosa kokkusurumise tõttu embrüonaalse arengu ajal. Kokkusurumine võib tekkida järgmistel põhjustel:
- Lokaliseeritud tihendite olemasolu emakas (tsüstid, armid, kasvajad).
- Mitmikrasedus.
Samuti võib loote lõualuu arengut häirida:
- Viirusnakkused, mida lapseootel ema raseduse ajal põdes.
- Neurotroofsed häired.
- Ebapiisav foolhappe kogus raseda naise kehas.
Pathogenesis
Pierre Robini sündroomi põhjustavad embrüonaalsed häired, mis on põhjustatud paljudest patoloogiatest sünnieelsel perioodil.
Pierre Robini sündroomi esinemist võib selgitada kolme patofüsioloogilise teooriaga.
Mehaaniline teooria: See teooria on kõige tõenäolisem. Alalõua aparaadi vähearenenud areng ilmneb 7. ja 11. rasedusnädala vahel. Keele kõrge asend suuõõnes viib suulaelõhede tekkeni, mille tõttu õõnesveen ei sulgu. See teooria selgitab klassikalist ümberpööratud U-kujulist lõhet ja sellega seotud huulelõhe puudumist. Oligohüdramnion võib etioloogias mängida rolli, kuna lootevee puudumine võib viia lõua deformatsioonini ja sellele järgneva keele kokkusurumiseni õõnesveenide vahele.
Neuroloogiline teooria: Neuroloogilise arengu hilinemist on täheldatud uvula ja neelukolonnide lihaste elektromüograafia ning maitsetundlikkuse abil, mis on tingitud hüpoglossaalnärvi juhtivuse hilinemisest.
Rombentsefaloni düsneuroregulatsiooni teooria: see teooria põhineb rombentsefaloni arengu häirimisel ontogeneesi ajal.
Lapse lõualuu alumise osa ebapiisav areng viib suuõõne märkimisväärse vähenemiseni. See omakorda põhjustab nn pseudomakroglossia, st keel nihkub neelu seina tahapoole. See patoloogia viib hingamisteede obstruktsiooni tekkeni.
Niikaua kui laps nutab või liigub, jäävad hingamisteed vabaks, kuid niipea kui laps magama jääb, tekib takistus uuesti.
Hingamisteede häirete tõttu on lapse toitmise protsess väga keeruline. Sel ajal tekib peaaegu alati hingamisteede obstruktsioon. Kui meditsiinilist korrektsiooni ei rakendata, võib selline patoloogia põhjustada kogu keha rasket ammendumist ja isegi surma.
Sümptomid Pierre Robini sündroom
Haigust iseloomustavad kolm peamist sümptomit:
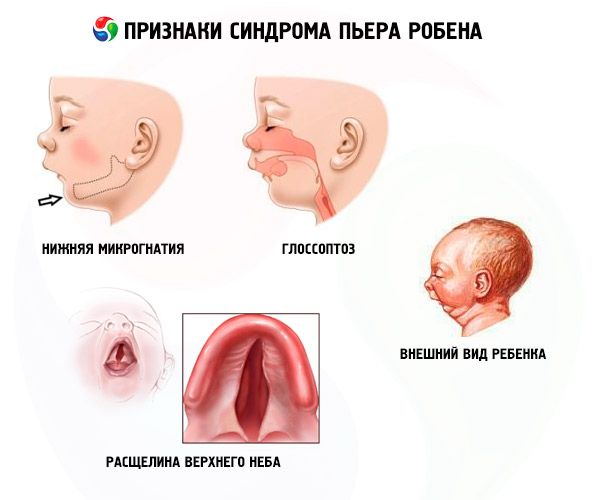
- Alumine mikrognaatia (alalõualuu alaareng, esineb 91,7% haiguse juhtudest). Seda iseloomustab alumise hambakaare taandumine 10-12 mm võrra ülemise kaare taha. Alumisel lõualuul on väike keha, nüri nurk. Laps saavutab normaalse arengu umbes 5-6-aastaselt.
- Glossoptoos (keele tagasitõmbumine selle ebapiisava arengu tõttu, täheldatud 70–85% juhtudest).
- Makroglossia ja anküloglossia on suhteliselt haruldased sümptomid, mida täheldatakse 10–15% juhtudest.
- Taevasse ilmub pragu.
- Bradüpnoe ja düspnoe.
- Kerge tsüanoos.
- Asfüksia, mis tekib kõige sagedamini lapse toitmise katsete ajal.
- Neelamine on võimatu või väga raske.
- Oksendamise tunne.
- Kõrva anomaaliad 75% juhtudest.
- Juhtiv kuulmislangus esineb 60% patsientidest, samas kui välise kuulmekanali atresia esineb ainult 5% patsientidest, ajalise luu mastoidõõne ebapiisav pneumaatika.
- Sisekõrva anomaaliad (külgmiste poolringikujuliste kanalite aplaasia, suur vestibulaarne akvedukt, kohlea karvarakkude kadu).
- Nina väärarengud on haruldased ja koosnevad peamiselt ninajuure anomaaliatest.
- Hambaväärarengud esinevad 30%-l juhtudest. Larüngomalatsia ja neelu-neelu puudulikkus esinevad ligikaudu 10–15%-l Pierre Robini sündroomiga patsientidest.
Pierre Robini sündroomi süsteemsed tunnused
Süsteemseid arenguanomaaliaid kirjeldatakse 10–85% registreeritud juhtudest.
Silmaanomaaliaid esineb 10–30%-l patsientidest. Nende hulka võivad kuuluda: kaugnägelikkus, lühinägelikkus, astigmatism, sarvkesta skleroos ja nina-pisarakanali stenoos.
Kardiovaskulaarsed patoloogiad: healoomulised südamekahinad, kopsuarteri stenoos, avatud arteriaalne juha, ovaalne aken, kodade vaheseina defekt ja pulmonaalne hüpertensioon. Nende levimus varieerub 5–58%.
Lihas-skeleti süsteemiga seotud anomaaliad (70–80% juhtudest): sündaktüülia, düsplastilised falangid, polüdaktüülia, klinodaktüülia, liigeste hüpermobiilsus ja ülajäsemete oligodaktüülia. Alajäsemete anomaaliad: jala anomaaliad (jalgsus, pöia adduktsioon), reieluu väärarengud (valgus- või varus-vaagen, lühikesed reieluud), puusa anomaaliad (kaasasündinud nihestus, kontraktuurid), põlveliigese anomaaliad (GENU VALGUS, sünkondroos). Selgroo väärarengud: skolioos, küfoos, lordoos, lülisamba düsplaasia, ristluu ja sabaluu siinuse agenees.
Kesknärvisüsteemi patoloogia: epilepsia, närvisüsteemi arengu hilinemine, hüdrotsefaalia. KNS-i defektide esinemissagedus on umbes 50%.
Kuseteede anomaaliad: laskumata munandid (25%), hüdronefroos (15%) ja hüdrokeel (10%).
Seotud sündroomid ja seisundid: Stickleri sündroom, trisoomia 11q sündroom, trisoomia 18, 4q deletsioonisündroom, reumatoidartropaatia, hüpokondroplaasia, Moebiuse sündroom.
Etapid
Haiguse raskusaste on kolm, mis sõltuvad lapse hingamisteede seisundist:
- Kerge - söötmisega on väiksemaid probleeme, kuid hingamine pole peaaegu keeruline. Ravi viiakse läbi ambulatoorselt.
- Mõõdukas – hingamine on mõõdukalt raskendatud, lapse toitmine on mõõdukalt raskendatud. Ravi viiakse läbi haiglas.
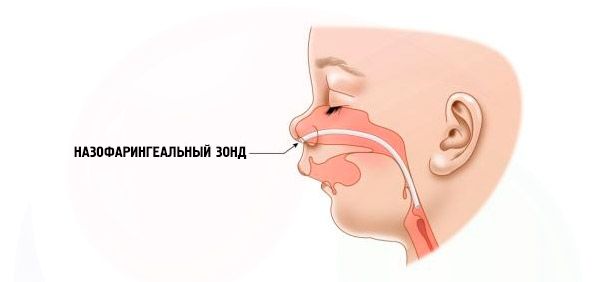
- Raske – hingamine on väga raskendatud, last ei saa normaalselt toita. Vajalik on kasutada spetsiaalseid seadmeid (ntranasaalne sond).
Tüsistused ja tagajärjed
Mikrognaatia ja glossoptoosi kombinatsioon võib põhjustada tõsiseid hingamisteede tüsistusi ja probleeme lapse toitmise ajal.
Pierre Robini sündroom põhjustab järgmisi tüsistusi:
- Stridoosne hingamine hingamisteede obstruktsiooni tõttu. Larüngomalaatsia või isegi uneasfüksia.
- Lapse psühhomotoorne areng jääb eakaaslastest oluliselt maha.
- Ka füüsiline areng jääb maha.
- Patsientide kõne on häiritud.
- Sagedased kõrvapõletikud, mis muutuvad krooniliseks ja põhjustavad kuulmislangust.
- Obstruktiivse uneapnoe sündroomi korral varieerub une ajal surma esinemissagedus 14–91% juhtudest.
- Probleemid hammastega.
Diagnostika Pierre Robini sündroom
Pierre Robini sündroomi diagnoosimine ei ole keeruline. See põhineb kliinilistel ilmingutel. Teiste patoloogiate välistamiseks on väga oluline konsulteerida geneetikuga.
Robini kaasasündinud anomaaliaga lastel on sünnist saati hingamisprobleemid, kuna keel vajub pidevalt tagasi. Beebi on rahutu, tema nahk on sinakas, sissehingamisel tuleb rinnast vilistavat hingamist. Söötmise ajal võib esineda lämbumist. Diagnoosi saab panna ka lapse ebatavalise välimuse - "linnu näo" - järgi. Sageli tekivad patsientidel muud defektid: lühinägelikkus, katarakt, urogenitaalsüsteemi patoloogia, südamepatoloogia, selgroo arengu anomaaliad.
Nende kliiniliste ilmingute põhjal ei ole spetsialistil raske õiget diagnoosi panna.
Kellega ühendust võtta?
Ravi Pierre Robini sündroom
Ravi viiakse läbi kohe pärast Pierre Robini sündroomiga lapse sündi. Kui haigus on kerge, on patsiendi seisundi parandamiseks vaja last pidevalt vertikaalselt või kõhuli hoida. Lapse pea peaks olema rinnale kallutatud. Söötmise ajal ei ole soovitatav last horisontaalasendis hoida, et toit ei satuks hingamisteedesse.
Kui alalõualuu arengupeetus on üsna väljendunud, kasutatakse kirurgilist sekkumist, et viia tagasitõmbunud keel normaalsesse füsioloogilisse asendisse. Rasketel juhtudel tõmmatakse keel üles ja fikseeritakse alumisele huulele. Väga rasketel juhtudel tuleb teha trahheostoomia, glossopeksia ja alalõualuu distraktsioonosteogenees.
Kasutatakse ka konservatiivset ravi.
Ravimid
Fenobarbitaal. Unerohi ja rahustav ravim, millel on krambivastane toime. Iga tablett sisaldab 100 ml fenobarbitaali. Annus on individuaalne, kuna see sõltub haiguse raskusest ja lapse seisundist. Ravim on keelatud maksapuudulikkuse, hüperkineesi, aneemia, müasteenia, porfüüria, suhkurtõve, depressiooni ja komponentide talumatuse korral. Selle võtmisel on võimalikud järgmised sümptomid: pearinglus, asteenia, hallutsinatsioonid, agranulotsütoos, iiveldus, madal vererõhk ja allergiad.
Klonasepaam. Epilepsia raviks välja kirjutatud ravim. Ravim sisaldab toimeainena klonasepaami, mis on bensodiasepiini derivaat. Sellel on krambivastane, anksiolüütiline ja lihaseid lõõgastav toime. Annuse määrab raviarst, kuid see ei tohiks ületada maksimaalset annust - 250 mikrogrammi päevas. Ärge võtke unetuse, lihashüpertoonia, psühhomotoorse agitatsiooni, paanikahäirete korral. Võtmisel on võimalikud järgmised sümptomid: letargia, iiveldus, düsmenorröa, peavalu, leukopeenia, uriinipeetus või -pidamatus, alopeetsia, allergia.
Sibazon. Saadaval lahuse ja rektaaltablettide kujul. Toimeaine on bensodiasepiini derivaat (sibazon). Sellel on rahustav, anksiolüütiline ja krambivastane toime. Annus on individuaalne. Kroonilise hüperkapnia, müasteenia ja bensodiasepiini talumatusega patsientidel on ravimi võtmine keelatud. Ravimi kasutamisel võivad tekkida järgmised sümptomid: iiveldus, kõhukinnisus, peavalu, pearinglus, luksumine, uriinipidamatus, allergiad.
Cortexini lüofilisaat. Nootroopse toimega ravim. Ravim sisaldab vees lahustuvate polüpeptiidfraktsioonide ja glütsiini kompleksi. Annus on individuaalne ja selle määrab raviarst vastavalt patsiendi seisundile. Patsientidel, kellel on korteksiini talumatus, on ravimi võtmine keelatud. Ravim võib põhjustada allergilisi reaktsioone.
Füsioteraapia
Tavaliselt kasutatakse sündroomi kergetes staadiumides asenditeraapiat, kus laps asetatakse kõhuli püstisesse asendisse, kuni gravitatsioon sunnib alumist lõualuu õigesti kasvama.
Kirurgiline ravi
Kirurgilist ravi kasutatakse peamiselt glossoptoosi korrigeerimiseks. On mitu meetodit:
- Keele toetamine hõbedase niidiga. Niit viiakse läbi igeme alumise osa ja alumise huule. Meetodit nimetatakse Douglase meetodiks.
- Duhameli meetod - patsiendi keele aluse ja mõlema põse kaudu viiakse läbi paks hõbedane niit. Kasutada mitte kauem kui kolmkümmend päeva.
- Ortopeedilised vahendid keele pikendamiseks ja fikseerimiseks.
- Üheaastaselt saab teha suulaelõhe korrigeerimise operatsiooni.
Prognoos
Haiguse prognoos ja kulg on rasked. Enamasti saabub surm esimestel elupäevadel haiguse mõõduka ja raske staadiumi korral (põhjuseks on lämbumine). Samuti on esimesel aastal surmaoht arvukate infektsioonide tõttu üsna kõrge.
Üle kahe aasta vanustel patsientidel on prognoos soodne.
 [ 36 ]
[ 36 ]