Artikli meditsiiniline ekspert

Uued väljaanded
Alkaptonuuria on kaasasündinud ensüümianomaalia.
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
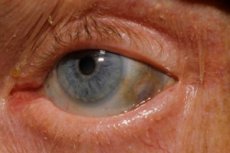
Üks väga haruldastest ainevahetushäiretest, alkaptonuuria, viitab aminohappe türosiini metabolismi kaasasündinud anomaaliatele.
Seda sündroomi võib nimetada ka homogentisaatoksüdaasi puudulikkuseks, homogentisinuuriaks, pärilikuks ochronoosiks või musta uriini haiguseks.[ 1 ]
Epidemioloogia
Statistika kohaselt ei esine alkaptonuuria juhtumeid rohkem kui üheksa miljoni inimese kohta. Ja enamikus Euroopa riikides on üks juhtum 100–250 tuhande elussünni kohta.
Euroopa riikide seas on erandiks Slovakkia (eriti suhteliselt väike loodepiirkond), kus alkaptonuuria levimus on üks juhtum 19 000 vastsündinu kohta. See on tõenäoliselt tingitud asjaolust, et seal elavate Slovakkia mustlaste perede seas on sugulusaretuse (nõbude vahelise abielu) tase Euroopa kõrgeim: 10–14%. [ 2 ]
Põhjused alkaptonuuria
Alkaptonuuria, aromaatse (homotsüklilise) α-aminohappe türosiini katabolismi (metaboolse lagundamise) kaasasündinud häire, täpsed põhjused on kindlaks tehtud: seda tüüpi ainevahetushäire on tingitud ühe tuhande geeni homosügootsetest või liitheterosügootsetest mutatsioonidest 3. kromosoomis, täpsemalt HGD geenist lookuses 3q21-q23 kromosoomi pikal harul. See geen kodeerib maksaensüümi homogentisaat-1,2-dioksügenaasi [ 3 ] (nimetatakse ka homogentisiinhappe oksüdaasiks või homogentisaatoksüdaasiks) nukleotiidjärjestusi – see on rauda sisaldav metalloproteiin, mis on vajalik türosiini lagundamise üheks etapiks organismis [ 4 ], [ 5 ].
Seega on alkaptonuuria ensüümi homogentisaat-1,2-dioksügenaasi defekt või täpsemalt selle geneetiliselt määratud puudulikkuse või täieliku puudumise tagajärg. [ 6 ]
Kuna alkaptonuuria on kaasasündinud ensüümipuudulikkus, pärandub see autosomaalselt retsessiivselt, see tähendab, et alkaptonuuria tekkeks lastel peab mõlemal vanemal olema ensüümi modifitseeritud geen, kuna kumbki neist annab lapsele edasi ainult ühe geeni koopia kahest olemasolevast.
Viimaste andmete kohaselt on HGD geeni modifikatsioonis üle kahesaja variandi ning kõige sagedamini täheldatakse missense-mutatsioone, translokatsiooni ja splaissingut.
Riskitegurid
Selle kaasasündinud ensümopaatia tekke ainus riskitegur on selle esinemine perekonnaanamneesis ja kahe HGD geeni modifitseeritud koopia pärilikkus, kui vanematel ei esine alkaptonuuriat (anomaalia edasikandumise risk on 25%) või kui ühel vanematest on see häire. [ 7 ]
Pathogenesis
Türosiin mängib võtmerolli valkude sünteesis, kromoproteiinide – nahapigmendi melaniini – tootmises, samuti kilpnäärmehormoonide ja katehhoolamiinide neurotransmitterite tootmises.
Türosiini hulga reguleerimise mehhanism rakkudes on väga keeruline ja organism normaliseerib selle liigset sisaldust selle lagundamise teel. Türosiini katabolismi protsess, nagu kõigi aromaatsete aminohapete puhul, on mitmeastmeline ja toimub mitmes etapis. Iga türosiini metaboolse lagundamise etapp toimub spetsiifilise ensüümi osalusel ja vaheühendi moodustumisel.
Seega lagundatakse aminohape kõigepealt para-hüdroksüfenüülpüruvaadiks, mis muundatakse alkaptooniks – 2,5-dihüdroksüfenüüläädikhappeks ehk homogentisiinhappeks. Seejärel peaks alkaptoon muunduma maleäädikhappeks, aga seda ei juhtu. [ 8 ]
Ja alkaptonuuria patogenees seisneb türosiini katabolismi biokeemiliste reaktsioonide lakkamises homogentisiinhappe moodustumise etapis: selle lagundamiseks pole lihtsalt vaja ensüümi – homogentisiinoksüdaasi.
Homogentisiinhapet organism ei kasuta ja see võib neerude kaudu eritumisel akumuleeruda. Lisaks oksüdeerub see bensokinoatsetaadiks (bensokinoonäädikhappeks), mis seondudes kudede ja kehavedelike molekulidega moodustab melaniini sarnase värvusega biopolümeerühendeid.
Nende vaheproduktide kogunemine koesse viib kõhrekoe kollageenistruktuuri häiruni, mis vähendab selle elastsust – koos alkaptonuuria paljude kliiniliste tunnuste ilmnemise ja tüsistuste tekkega.
Sümptomid alkaptonuuria
Alkaptonuuriat vastsündinutel ja imikutel iseloomustab uriini tumenemine. Õhuga kokkupuutel muutub mähkmetel, paberrätikutel ja aluspesul olev uriin tumepruuniks; see on tingitud homogentisiinhappe kogunemisest ja vabanemisest, mis oksüdeerub bensokinoatsetaadiks. [ 9 ]
Muude sümptomite puudumisel ei diagnoosita alkaptonuuriat väikelastel sageli õigeaegselt, kuna uriin võib pärast mitmetunnist urineerimist tumeneda. Mõnede andmete kohaselt diagnoositakse kliinilistes tingimustes vaid viiendikul alla 12 kuu vanustest lastest, kes on sündinud selle ensüümi puudulikkusega. Seetõttu on väga oluline, et vanemad pööraksid tähelepanu oma imikute eest hoolitsemisele.
Lisaks on varajaste nähtude hulka silmade kõvakesta ning kõrvade ja nina kõhrede pigmentatsioon (sinakashall värvus), mida sageli nimetatakse ochronoosiks.[ 10 ]
Aja jooksul ilmnevad muud sümptomid:
- naha tugev pigmentatsioon põsesarnadel, kaenlaalustel ja suguelunditel;
- riiete värvimine kokkupuutel keha higiste piirkondadega;
- üldise nõrkuse rünnakud;
- kähe hääl.
Tuleb meeles pidada, et alkaptonuuria ja ochronosis, nagu eespool märgitud, on türosiini katabolismi sama häire sünonüümsed nimetused.
Vahtrasiirupi uriinihaigus ja alkaptonuuria. Kaasasündinud vahtrasiirupi uriinihaigus ehk leukinoos on samuti ainevahetushäire, millel on sama pärilikkusmuster ja isegi mutatsioonid esinevad samas kromosoomis, kuid mõjutavad hargnenud ahelaga α-ketohappe dehüdrogenaasi ensüümikompleksi kodeerivat geeni. Seetõttu ei suuda organism lagundada teatud valkude komponente, eriti aminohappeid leutsiini, isoleutsiini ja valiini. Selle haiguse korral on uriinil (ja kõrvavaigul) magus lõhn; lisaks hõlmab seda tüüpi orgaanilise atsideemia kliiniline pilt hüpopigmentatsiooni, vererõhu kõikumisi, krampe, oksendamist ja kõhulahtisust, veresuhkru taseme langust, ketoatsidoosi, hallutsinatsioone jne. Laste suremus on üsna kõrge; täiskasvanutel võib ravita ajuödeemi tõttu tekkida kooma ja surm.
Albinismi ja alkaptonuuriat „ühendab“ ainult türosiin. Albinism, sealhulgas okulokutaanne albinism, on põhjustatud geneetilistest mutatsioonidest, mis mõjutavad pigmendi melaniini tootmist. Kaasasündinud muutusi on täheldatud TYR geenis 11. kromosoomis (11q14.3), mis kodeerib türosinaasi, vaske sisaldavat melanosoomi ensüümi, mis on vajalik nahapigmendi moodustumiseks türosiini metabolismi produktide põhjal. See haigus on palju levinum kui alkaptonuuria.
Tüsistused ja tagajärjed
Türosiini vaheühendite metaboliitide – homogentisiin- ja bensokinoonäädikhapete – toimel ilmnevad alkaptonuuria tagajärjed ja tüsistused reaktiivsete pigmenteeritud polümeeride ladestumise, kollageenifibrillide hävimise ja kõhre seisundi halvenemise (koos nende mehaanilise stressi vastupidavuse vähenemisega) tõttu.
Aastate jooksul täiskasvanueas tekivad suurte liigeste (puusa-, ristluu- ja niudeluu- ning põlveliigese) degeneratiivne artriit ja osteoartriit; lülidevahelised ruumid ahenevad (eriti nimme- ja rindkerelülides) – koos kaltsifikatsiooni ja osteofüütide moodustumisega; subhondraalsete luuplaatide koe tihedus väheneb ja alusluud võivad läbida patoloogilise ümberkujunemise koos kasvajate ja deformatsioonide tekkega. [ 11 ]
Sama kaltsifikatsiooni tõttu võivad esineda südameklappide (aordi- ja mitraalklapi) ning pärgarterite kahjustused – koos südame isheemiatõve tunnustega, samuti neerukivide ja eesnäärme moodustumine. [ 12 ], [ 13 ]
Diagnostika alkaptonuuria
Tavaliselt põhineb kaasasündinud ainevahetushäirete diagnoosimine keha bioloogiliste vedelike uurimisel.
Milliste testide ja reaktsioonide põhjal saab alkaptonuuriat diagnoosida? Uriinianalüüsid on vajalikud homogentisiinhappe tuvastamiseks ja selle taseme määramiseks (normaalne – 20–30 mg päevas, kõrgenenud – 3–8 g). Uriiniproovi uuritakse gaasikromatograafia või massispektromeetria abil, kasutades vedelikkromatograafiat; võimalik on teha sõeltest raudkloriidi olemasolu kohta uriinis. [ 14 ]
Samuti on olemas kiirdiagnostika meetod – alkaptooni määramine kuivanud uriiniplekkides paberil (värvi intensiivsuse järgi).
Diagnoosi selgitamisel hõlmab instrumentaalne diagnostika (radiograafia) osteoartriidi ja teiste liigesepatoloogiate radioloogiliste tunnuste tuvastamist patsientidel.
Diagnoosi kinnitavad pärilike haiguste diagnoosimise molekulaargeneetilised meetodid, näiteks geneetiline testimine ja DNA sekveneerimine. [ 15 ]
Diferentseeritud diagnoos
Diferentsiaaldiagnoos hõlmab vastsündinu hemokromatoosi ja ägedat maksapuudulikkust, melaninuuriat, ägedat vahelduvat porfüüriat, hemofagotsütaarset lümfohistiotsütoosi, primaarset mitokondriaalset patoloogiat, reumatoidartriiti, anküloseerivat spondüliiti.
Kellega ühendust võtta?
Ravi alkaptonuuria
Alkaptonuuria peamine ravi on askorbiinhappe suurte annuste (vähemalt 1000 mg päevas) suukaudne manustamine. Lastel suurendab see homogentisiinhappe eritumist uriiniga ja täiskasvanutel vähendab selle derivaadi, bensokinoonäädikhappe, sisaldust uriinis ning aeglustab selle seondumist liigeste sidekoe struktuuride ja kollageeniga. [ 16 ]
Lääne-Euroopa kliinikud testivad ravimit Nitisinoon (Orfalin), mis kuulub metaboliitide rühma ja pärsib türosiini katabolismi teist etappi: para-hüdroksüfenüülpüruvaadi muundumist homogentisiinhappeks. Selle farmakoloogilise aine kasutamine viib aga türosiini akumuleerumiseni ja võib põhjustada tõsiseid kõrvaltoimeid, sealhulgas sarvkesta hägustumist ja valguskartust, nina- ja maoverejooksu, maksapuudulikkust, muutusi veres jne. Sellest hoolimata on Ameerika Ühendriikides Nitisinoon FDA poolt heaks kiidetud I tüüpi türosineemia raviks. [17 ], [ 18 ]
Seega rakendatakse alkaptonuuria põhjustatud liigeseprobleemide korral füsioteraapiat – treeningravi lihasjõu suurendamiseks ja liigeste liikuvuse parandamiseks, balneoteraapiat ja peloidteraapiat valu leevendamiseks.
Kuigi türosiini ei saa organism mitte ainult toiduga, vaid seda toodetakse ka ise, soovitatakse alkaptonuuriaga patsientidel järgida madala valgusisaldusega dieeti ja piirata türosiinirikaste toitude, eelkõige veise- ja sealiha, piimatoodete (eriti juustude), kaunviljade, pähklite ja seemnete tarbimist.
Ärahoidmine
Geenimutatsioonide ennetamine on võimatu, kuid kaasasündinud häirete suure riskiga laste sünni vältimiseks on olemas meditsiiniline geneetiline nõustamine, mis on vajalik enne planeeritud rasedust neile paaridele, kelle perekonnas on esinenud pärilikke haigusi. [ 19 ]
Prognoos
Alkaptonuuria surmaga lõppevad tagajärjed on väga haruldased ning surma võivad põhjustada tõsised südame ja neerudega seotud tüsistused. Seega on alkaptonuuriaga inimeste üldine eluiga hea.
Kuid elukvaliteeti vähendab intensiivne valu liigestes või selgroos, millega kaasneb märkimisväärne liikuvuse piiramine, mis on sageli progresseeruv.