Trigonotsefaalia
Viimati vaadatud: 07.06.2024

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
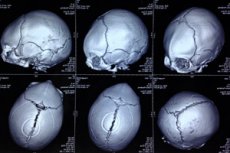
Kaasasündinud anomaalia kolju deformatsiooni kujul, milles imikute pead on ebakorrapärase kujuga ja kolju näib kolmnurkset määratletud kui trigonocephaly (Kreeka trigononist - kolmnurgast ja kephale - peast). [1]
Epidemioloogia
Kraniosünostoosi esinemissagedus on hinnanguliselt umbes viis juhtumit 10 000 elussünni kohta (või üks juhtum elanikkonnas 2000–2 500 kohta). [2]
Kraniosünostoos on juhuslik 85% -l juhtudest, ülejäänud juhtumid toimuvad sündroomi osana. [3]
Statistika kohaselt on mediaalse eesmise õmbluse enneaegne sulandumine kraniosünostoosi teine levinum vorm ja trigonocephaly moodustab ühe juhtumi 5000–15 000 vastsündinu kohta; Selle anomaaliaga meessoost imikute arv on peaaegu kolm korda suurem kui naissoost imikutel. [4]
Umbes 5% juhtudest esineb see kaasasündinud anomaalia perekonna ajaloos. [5]
Põhjused trigonotsefaalia
Kolju normaalne moodustumine toimub primaarse kasvu ja luude ümberehituse keskpunktide olemasolu tõttu-kraniofaciaalsed sünaartoosid (liigendused), mis pea skeleti arendamine suletakse teatud ajal, pakkudes luude sulandumist. [6]
Vastsündinu kolju eesmine luu (OS Frontale) koosneb kahest poolest, mille vahel on vertikaalne kiuline ühendus-mediaalne esi- või metopiline õmblus (Kreeka Metoponist-laubast), mis kulgeb nina keskjoone ülaosast ülespoole eesmise eesliinist ülespoole. See on ainus kiuline kraniaalõmblus, mis imikueas üle kasvab: 3-4 kuud kuni 8-18 kuuni. [7]
Vaata ka. - kolju muutub pärast sündi
Trigonocephaly põhjused on metopiline kraniosünostoos (kraniostenoos) või metopiline sünostoos (Kreeka sünkroonist koos ja osteon-luu), s.o enneaegne (enne kolmandat kuud enne kolmandat kuud) immobilefusioone, mis on mõeldud lambaluude lauale. Seega on kraniosünostoos ja trigonotsefaalia seotud kui põhjus ja tagajärg või patoloogiline protsess ja selle tulemus. [8]
Enamikul juhtudel on lapse trigonotsefaalia primaarse (isoleeritud) kraniosünostoosi tagajärg, mille täpne põhjus pole teada. Isoleeritud kraniosünostoos toimub juhuslikult, tõenäoliselt geneetiliste ja keskkonnategurite kombinatsioonide tõttu. [9]
Kuid trigonotsefaalia võib olla osa kaasasündinud sündroomidest, mis tulenevad erinevate geenide kromosomaalsetest kõrvalekalletest ja mutatsioonidest. Nende hulka kuulub: Opitzi trigonocephaly sündroom (igav-opitz-sündroom), aper sündroom, loeys-dietzi sündroom, Pfeiffer Syndrom, Jackson-Weissi sündroom, Craniofacial Dysostoos või düstrom , Setre-Chotzen, Muenke sündroomid. Sellistel juhtudel nimetatakse trigonocephalyt sündroomiliseks trigonocephalyks. [10]
Sündides on aju suurus tavaliselt 25% selle täiskasvanute suurusest ja esimese eluaasta lõpuks ulatub see umbes 75% -ni täiskasvanu ajust. Kuid aju primaarse kasvu aeglustumisega on võimalik niinimetatud sekundaarne kraniosünostoos. Viivituse etioloogia on seotud metaboolsete häirete, mõningate hematoloogiliste haiguste, teratogeensete mõjudega kemikaalide lootele (sealhulgas farmaatsia koostises). [11]
Ekspertide sõnul püsib kogu elu vältel täiskasvanute trigonotsefaalia täiskasvanutel, keda ei ravitud isoleeritud kraniosünostoosi või kaasasündinud sündroomi tagajärjel. [12]
Riskitegurid
Spetsialistid peavad geneetilisteks trigonotsefaalia (ja metopilise kraniosünostoosi) peamisi riskifaktoreid: viimase kahe aastakümne jooksul on tuvastatud enam kui 60 geeni, mille mutatsioonid on seotud imikute kraniaalsete luude enneaegse liikumisega.
Suurenenud on kraniofaciaalse sünatroosi ja üldiste osteogeneesi (luude moodustumise) kõrvalekalde oht loote väärarengu, emakasisese hüpoksia, mitmete raseduste, alkoholi, narkootikumide kasutamise või suitsetamise ajal. [13]
Pathogenesis
Valitseva teooria kohaselt seisneb trigonotsefaalia patogenees loote osteogeneesis halvenenud raseduse ajal, mis on enamasti põhjustatud geneetilistest teguritest, kuna vastsündinutel tuvastatakse juhuslikud kromosomaalsed kõrvalekalded metopilise kraniosünostoosiga. Näiteks on trisoomia 9P üks sagedasemaid, põhjustades kraniofacial ja skeleti defekte, vaimseid ja psühhomotoorseid arengu viivitusi. [14]
Mediaalse eesmise õmbluse liiga varase sulandumise tõttu on selle kolju piirkonna kasv keeruline: eesmise luu külgmine kasv on piiratud kraniaalse fossa lühenemisega; Otsaesise keskjoonel moodustatakse kondine katuseharja; Silma orbiidi moodustavate luude ja ajaliste luude depressiooni moodustavate luude lähenemine on olemas. [15]
Kuid kolju kasv teistes piirkondades jätkub: kolju tagumise osa kompenseeriv sagitaal (anteroposterior) ja põiksuunaline kasv (selle parieto-kuklaluu laienemisega), aga ka näo ülemise osa vertikaalne ja sagitaalne kasv. Nende kõrvalekallete tagajärjel omandab kolju ebaregulaarse kuju - kolmnurkse kuju.
Sümptomid trigonotsefaalia
Trigonotsefaalia peamised sümptomid on pea kuju ja välimuse muutused:
- Pea ülaosast vaadates on kolju kolmnurkse kujuga;
- Kitsas otsmik;
- Silmapaistev või palpeeritav katuseharja (kondine väljaulatuvus), mis kulgeb piki otsaesise keskelt, mis annab eesmise luu terava (keelustatud) kuju;
- Silmapesade (supraorbitaalsete servade tasandamine) ja hüpotelorism (silmade vaheline kaugus) deformatsioon.
Eesmine (eesmine) fontanelle võib samuti enneaegselt suletud olla.
Sündroomse trigonocephaly puhul on ka muid anomaaliaid ja märke vaimne alaareng lastel. [16]
Tüsistused ja tagajärjed
Diagnostika trigonotsefaalia
Diferentseeritud diagnoos
Diferentsiaaldiagnostika on vajalik, et eristada sündroomsefekti isoleeritud metopilisest sünostoosist, millele lapsele antakse genotüübi testimine.
Ravi trigonotsefaalia
Mõnel lapsel on metopilise sünostoosi juhtumid üsna kerged (kui otsmikul on ainult märgatav vagu ja muid sümptomeid pole), mis ei vaja konkreetset ravi. [21]
Raske trigonotsefaalia ravi on kirurgiline ja koosneb operatsioonist, et korrigeerida pea kuju ja võimaldada aju normaalset kasvu, samuti näo luu deformatsioonide kirurgilist parandust. [22]
See kirurgiline sekkumine - metopiline õmbluse sünostektoomia, orbitaalmarginaali nihe ja kranioplastika - viiakse läbi enne 6-kuulist vanust. Last jälgitakse kuni ühe aasta vanuseni; Esimese paari eluaasta jooksul uuritakse last perioodiliselt veendumaks, et kõne-, motoorseid ega käitumisprobleeme pole. [23]
Ärahoidmine
Selle sünnidefekti ennetamise meetodeid ei ole välja töötatud, kuid geneetiline nõustamine võib vältida lapse sündi ravimatu kraniotserebraalse patoloogiaga.
Ja loote kraniosünostoosi saab tuvastada selle pea sünnieelse ultraheli abil raseduse teisel ja kolmandal trimestril.
Prognoos
Prognoos sõltub suuresti kolju deformatsiooni astmest, mis mõjutab aju neurokognitiivseid funktsioone. Ja kui korrigeerivat operatsiooni ei tehta, on trigonotsefaaliaga lastel - võrreldes tervete eakaaslastega - üldine üldised kognitiivsed võimed, kõne, nägemine, tähelepanu ja käitumisprobleemid.
Использованная литература