Artikli meditsiiniline ekspert

Uued väljaanded
Angelmani sündroom lastel ja täiskasvanutel
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.

On mitmeid haigusi, mille puhul väljendid nagu "hoolitse enda eest ja sa ei jää haigeks" kõlavad vähemalt naeruväärselt. Need on patoloogiad, mille puhul on lapse kehas juba enne sündi olemas mõned vaimsed ja füüsilised kõrvalekalded, kuid vanemad ei ole selles süüdi. Sellised haigused on põhjustatud kromosoomide mutatsioonidest või kõrvalekalletest ja neid nimetatakse kromosomaalseteks ehk geneetilisteks. Angelmani sündroom, Downi sündroom, Patau sündroom, Edwardsi sündroom, Turneri sündroom, Prader-Willi sündroom - see on vaid osa geneetilistest haigustest üsna korralikust loetelust.
Õnneliku mehe sündroom
Seekord räägime patoloogiast, mis sai nime inglise lastearsti Harry Angelmani järgi. Angelman tõstatas selle probleemi esmakordselt 1965. aastal, olles eelmisel päeval oma praktikas kohanud kolme ebatavalist last, keda ühendasid ühised omapärased sümptomid. Arst nimetas neid lapsi nukulasteks ja kirjutas nende kohta artikli, mis algselt kandis pealkirja "Lapsed-marionetid". Artikkel ise ja selle pealkiri kirjutati Verona muuseumides nähtud maali ajendil. Maal kujutas naeravat poissi ja seda nimetati "Nukupoisiks". Maalil kujutatud lapse seos kolme lapsega, keda Angelman kunagi oma praktikas kohtas, ajendas lastearsti lapsed haiguse tõttu ühte rühma ühendama.
Pole midagi üllatavat selles, et artiklis mainitud lapsi teised arstid ei märganud. Lõppude lõpuks tundus esmapilgul, et neil on täiesti erinevad haigused, nii erinev oli haiguse üldine kliiniline pilt kolmel erineval juhul. Võib-olla oleks "uus" kromosomaalne patoloogia huvitanud ka teisi teadlasi, kuid sel ajal polnud geneetika veel piisavalt arenenud, et kinnitada inglise arsti hüpoteesi. Seetõttu visati artikkel pärast teatud huvi tekkimist selle vastu pikaks ajaks tagariiulile.
Järgmine mainimine Angelmani sündroomist, milleks nimetati nüüd inglise lastearsti G. Angelmani artiklit, pärineb 20. sajandi 80. aastate algusest. Ja alles 1987. aastal õnnestus leida põhjus, miks väike osa lastest sünnib selliste kõrvalekalletega, et väljastpoolt näivad nad pidevalt naeratavat ja õnnelikku. Tegelikult pole see üldse tõsi ja naeratus on vaid grimass, mille taga peidab end õnnetu inimhinge ja vanemate valu.
Epidemioloogia
Statistika kohaselt võib lapse kromosomaalne mutatsioon areneda nii vanemate sarnaste mutatsioonide taustal kui ka nende puudumisel. Angelmani sündroomil (AS) ei ole selget pärilikku olemust, kuid kromosomaalsete mutatsioonidega vanematel on patoloogia tekkimise tõenäosus üsna suur.
Samuti on huvitav, et kui peres on juba AS-iga laps, on ühe protsendi tõenäosus saada teine laps sama häirega, isegi kui vanemad on terved.
Angelmani sündroomiga patsientide arvu kohta täpset statistikat endiselt pole. Võib-olla on põhjuseks sümptomite mitmekesisus, mis võivad esineda teatud koosseisus või pikka aega üldse mitte esineda. Eeldatakse, et haiguse levimus on: 1 laps 20 000 vastsündinu kohta. Kuid see arv on väga ligikaudne.
Põhjused Angelmani sündroom
Angelmani sündroom on kromosomaalse patoloogia meditsiiniline nimetus, kuid see pole kaugeltki ainus. Inimesed kutsuvad seda haigust nukulaste sündroomiks, õnneliku nuku sündroomiks, Petruška sündroomiks ja naerava nuku sündroomiks. Inimesed mõtlevad välja igasuguseid nimesid (mõnikord isegi solvavaid nii patsientidele endile kui ka nende vanematele), aga haigus on haigus, ükskõik kui naljakas see ka välja ei näeks ja millised on selle põhjused.
Ja Angelmani sündroomi, nagu paljude teiste geneetiliste patoloogiate, tekke põhjused on kõigil juhtudel ühe kromosoomi või kogu kromosoomikomplekti struktuuri häired. Kuid meie puhul peitub kogu probleem emalt edasi antud 15. kromosoomis. See tähendab, et isapoolsel kromosoomil pole sel juhul kõrvalekaldeid, kuid naissoost kromosoomis esinevad teatud mutatsioonid.
Kromosomaalse anomaalia tüübi järgi liigitatakse Angelmani sündroom kromosomaalseks mutatsiooniks. Selliseid mutatsioone peetakse:
- Deletsioon (teatud geenide komplekti sisaldava kromosoomiosa puudumine; kui üks geenidest puudub, räägime mikrodeletsioonist), mis tekib kahe katkemise ja ühe taasühinemise tagajärjel, kui algse kromosoomi osa kaob.
- Duplikatsioon (kromosoomis oleva täiendava sektsiooni olemasolu, mis on olemasoleva koopia), mis enamasti viib inimese surmani ja harvemini viljatuseni.
- Inversioon (kromosoomi ühe osa pööramine 180 kraadi võrra ehk vastassuunas ja seejärel paiknevad selles olevad geenid vastupidises järjekorras), kui kromosoomi katkenud otsad on ühendatud algsest erinevas järjekorras.
- Insertsioon (kui osa kromosoomi geneetilisest materjalist on paigast ära),
- translokatsioon (kui teatud kromosoomi osa on kinnitatud teise kromosoomi külge; selline mutatsioon võib olla vastastikune ilma sektsioonide kadumiseta).
Saades pahaaimamatult emalt muteerunud kromosoomi, on laps määratud sündima kõrvalekalletega. Angelmani sündroomi kõige levinumaks põhjuseks peetakse endiselt ema 15. kromosoomi deletsiooni, kui väike lõik puudub. Harvemini esinevateks mutatsioonideks "naerva nuku" sündroomis peetakse:
- ümberpaigutamine
- unipaternaalne disoomia (kui laps sai isalt kromosoomipaari, siis ema kromosoom puudub),
- DNA geenide mutatsioon, mis on nii peamine ehitusmaterjal (geneetiline materjal) kui ka juhised selle õigeks kasutamiseks (eriti ube3a geeni mutatsioon ema kromosoomis).
Ühe nimetatud mutatsiooni esinemine vanematel on Angelmani sündroomi tekke riskitegur lastel. Kuid mitte ainult kromosomaalsed mutatsioonid, vaid ka genoomsed mutatsioonid (mis on seotud kromosoomikomplektide kvantitatiivse muutusega ja on sagedasemad kui kromosomaalsed mutatsioonid) võivad provotseerida haiguse arengut lapsel. Levinud genoomsete mutatsioonide hulka kuulub kromosoomitrisoomia (kui inimese kromosoomikomplektis on rohkem kui 46 kromosoomi).
Selleks, et lapsel patoloogia ilmneks, ei ole vanematel üldse vaja kromosomaalseid kõrvalekaldeid. Ja ometi on teatud protsent patsiente, kelle haigus on pärilik.
Pathogenesis
Sukeldume veidi sügavamale bioloogiasse ehk täpsemalt geneetikasse. Iga üksiku inimorganismi geneetiline informatsioon sisaldub 23 kromosoomipaaris. Üks kromosoom paarist pärineb lapsele isalt, teine emalt. Kõik kromosoomipaarid erinevad kuju ja suuruse poolest ning kannavad endas teatud informatsiooni. Seega vastutab 23. kromosoomipaar (X- ja Y-kromosoomid) lapse sugutunnuste kujunemise eest (XX - tüdruk, XY - poiss, Y-kromosoomi saab laps aga ainult isalt).
Ideaalis saab laps vanematelt 46 kromosoomi, mis moodustavad tema geneetilised omadused, määrates ta indiviidina. Suuremat kromosoomide arvu nimetatakse trisoomiaks ja seda peetakse normist kõrvalekalleks. Näiteks 47. kromosoomi olemasolu kromosoomikomplektis (karüotüüp, mis määrab liigi ja individuaalsed omadused) põhjustab Downi sündroomi esinemist.
Kui kromosoome värvida spetsiaalse värvainega, siis mikroskoobi all on igaühel neist näha erinevat tooni triipe. Iga triibu sees on tohutu hulk geene. Kõik need triibud on teadlaste poolt nummerdatud ja neil on kindel asukoht. Ühe triibu puudumist peetakse normist kõrvalekalleks. Angelmani sündroomi korral võib väga sageli täheldada emakromosoomi segmentide puudumist vahemikus q11-q13, mis asuvad pikas harus ja milles on vaid umbes 4 miljonit DNA alust.
Kromosoomi põhikomponendiks peetakse uskumatult pikka DNA molekuli, mis sisaldab tuhandeid geene ja kümneid ning sadu miljoneid lämmastikaluseid. Seega sisaldab Angelmani sündroomi ja mitmete teiste haiguste tekke eest vastutav 15. kromosoom 1200 geeni ja umbes 100 miljonit alust. Igasugused DNA molekuli struktuuri häired mõjutavad kindlasti tulevase lapse välimust ja arengut.
Geenides sisalduv geneetiline teave muundatakse valguks või RNA-ks. Seda protsessi nimetatakse geeniekspressiooniks. Sel viisil saab vanematelt saadud geneetiline teave nii vormi kui ka sisu, mis kehastub nende ainulaadses nais- või meespärijas.
Mitteklassikalise pärimistüübiga on mitmeid patoloogiaid, sealhulgas Angelmani sündroom, mille puhul vanematelt paariskromosoomide osana saadud geenid kannavad vanemate ainulaadset jäljendit ja avalduvad erineval viisil.
Seega on Angelmani sündroom silmatorkav näide genoomsest imprintingust, kus geeniekspressioon lapse kehas sõltub otseselt sellest, milliselt vanemalt alleelid pärinesid (ühe geeni erinevad vormid, mis on saadud isalt ja emalt ning paiknevad paariskromosoomide identsetes osades). See tähendab, et sündroomi tekkeni viivad ainult ema kromosoomi anomaaliad, samas kui isa kromosoomi mutatsioonid ja struktuurihäired põhjustavad täiesti erinevaid patoloogiaid.
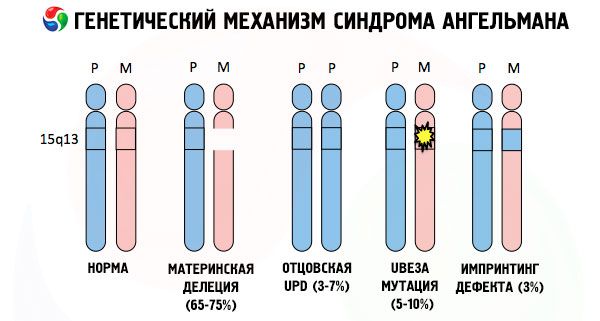
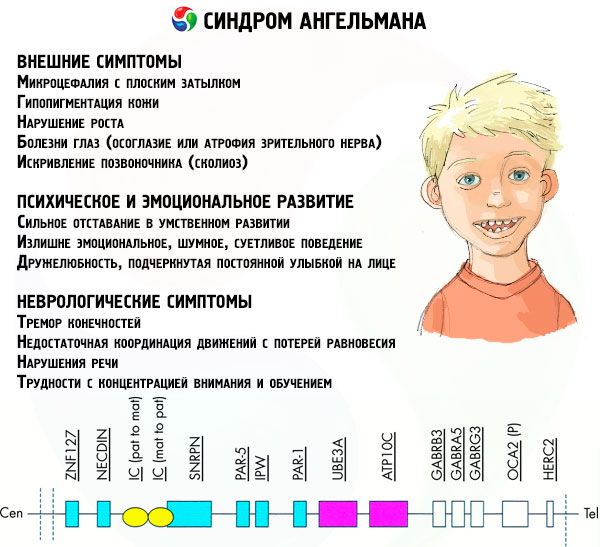
Selle patoloogia korral puudub ema kromosoomis teatud geene või on üksikute geenide aktiivsus kadunud/vähenenud (valdaval enamusel juhtudel on tegemist ube3a geeniga, mis osaleb ubikvitiini metabolismis – valk, mis reguleerib teiste valkude lagundamist). Selle tulemusena diagnoositakse lapsel vaimse arengu kõrvalekalded ja füüsilised deformatsioonid.
Sümptomid Angelmani sündroom
Angelmani sündroomi sümptomid mõjutavad lapse elu ja arengu erinevaid aspekte: füüsilist, neuroloogilist, vaimset. Selle põhjal saab tuvastada 3 sümptomite rühma, mis viitavad selle patoloogia arengule.
- Välised ehk füüsilised sümptomid:
- ebaproportsionaalselt väike pea võrreldes normaalse suurusega keha ja jäsemetega,
- liiga lai suu,
- näol on peaaegu alati naeratus (avatud suuga),
- hõredad hambad,
- kitsas ülahuul,
- sageli väljaulatuv lai keel,
- väljaulatuv alumine lõualuu,
- terav lõug,
- väga hele nahk, sageli karvad (albinism, mis on seotud asjaoluga, et keha ei tooda pigmendi melaniini),
- tumedad laigud heledal nahal (hüpopigmentatsioon ebapiisava melaniini tootmise tõttu)
- füüsilised või välised sümptomid: silmahaigused, näiteks strabismus või nägemisnärvi atroofia,
- selgroo kõverus (skolioos),
- jäigad jalad (kõndides ei painuta inimene liigeste madala liikuvuse tõttu jalgu põlvedes, seega võrdlus nuku kõnnakuga).
- Vaimse ja emotsionaalse arenguga seotud sümptomid:
- raske vaimne alaareng,
- liialt emotsionaalne, lärmakas ja rahutu käitumine,
- sagedane käte plaksutamine,
- väljendatud sõbralikkus, mida rõhutab pidev naeratus näol,
- sagedane naer ilma põhjuseta.
- Neuroloogilised sümptomid:
- jäsemete värisemine,
- liigutuste ebapiisav koordineerimine koos tasakaalu kaotusega,
- vähenenud lihastoonus,
- mitmesugused unehäired,
- sagedased hüsteerilised hood lapsepõlves,
- kõnehäired (laps hakkab hilja rääkima, tal on halvad suhtlemisoskused ja ebaselge kõne),
- hüperaktiivsus suurenenud erutuvuse taustal,
- raskused keskendumisel ja õppimisel.
Kuid see on haiguse üldistatud pilt. Tegelikult sõltub Angelmani sündroomi kliiniline pilt suuresti haiguse arenguetapist ja patoloogiat põhjustanud kromosomaalse mutatsiooni tüübist. See tähendab, et haiguse sümptomid võivad erinevatel patsientidel oluliselt erineda, mis pikka aega ei võimaldanud meil eristada patoloogiat teistest sarnase kliinilise pildiga patsientidest.
Sümptomite koguarvust võime esile tõsta neid, mis on iseloomulikud kõigile patsientidele ilma eranditeta:
- raske vaimne alaareng,
- sobimatu käitumine (ebamõistlik naer, suurenenud erutuvus, halb keskendumisvõime, eufooria),
- motoorsete oskuste vähene areng,
- liigutuste halb koordinatsioon, kõnnaku ataksia (ebaühtlane tempo, küljelt küljele kõikumine jne), jäsemete treemor.
- kõne arenguhäire, kus domineerivad mitteverbaalsed suhtlusvahendid.
Enamiku patsientide sümptomite hulgast saab eristada järgmisi:
- pea ja keha ebaproportsionaalsus, mis on põhjustatud füüsilise arengu hilinemisest;
- paljudel patsientidel on kolju kuju selline, et aju suurus jääb väiksemaks kui tervetel inimestel (mikrotsefaalia),
- epilepsiahoogude esinemine enne 3. eluaastat, mille tugevus ja sagedus vanemas eas järk-järgult väheneb,
- EEG parameetrite moonutamine (madala sagedusega lainete kõikumised ja suur amplituud).
Need sümptomid on üsna levinud, kuid 20%-l Angelmani sündroomiga patsientidest neid ei esine.
Veelgi harvemini on võimalik diagnoosida selliseid haiguse ilminguid nagu:
- raske või kerge strabismus,
- keele liikumise halb kontroll, mille tagajärjel ajavad patsiendid oma keele sageli ilma põhjuseta välja,
- raskused neelamise ja imemisega, eriti väikelastel,
- naha ja silmade pigmentatsiooni häired,
- kõndimise ajal käed üles tõstetud või painutatud,
- hüperrefleksia
- unehäired, eriti lapsepõlves,
- sagedane süljeeritus,
- kustutamatu janu,
- liiga aktiivsed närimisliigutused,
- ülitundlikkus kuumuse suhtes,
- lame pea tagaosa,
- väljaulatuv alumine lõualuu,
- siledad peopesad.
Üsna suurel protsendil patsientidest on probleeme urineerimisega, mida nad halvasti kontrollivad, halvenenud peenmotoorika, mis tekitab raskusi enesehoolduses ja õppimises, ning liigne kaal. Peaaegu kõik patsiendid kogevad puberteeti hiljem kui terved eakaaslased.
Angelmani sündroomiga lapsed tajuvad suulist kõnet hästi ja saavad sellest aru, kuid ei taha vestluses osaleda, piirdudes oma kõnes mitmekümne igapäevaelus vajaliku sõnaga. Täiskasvanueas näevad sellised patsiendid aga geneetiliste patoloogiateta eakaaslastest nooremad välja.
Paljud Angelmani sündroomi sümptomid on ebapüsivad, seega muutub haiguse kliiniline pilt vanusega oluliselt. Krambid ja epilepsiahood muutuvad harvemaks või kaovad täielikult, patsient muutub vähem erutuvaks ja uni paraneb.
Tüsistused ja tagajärjed
Angelmani sündroom on raske, praegu praktiliselt ravimatu kromosomaalne patoloogia, mis jätab patsiendid ilma võimalusest elada normaalset elu. See, milline saab olema AS-iga lapse elu, sõltub suuresti kromosomaalse anomaalia tüübist.
Kromosoomi segmendi dubleerimine on enamikul juhtudel eluga kokkusobimatu. Ja isegi kui sellised patsiendid ei sure imikueas ja ei jõua puberteedieasse, pole neil võimalust lapsi saada.
Angelmani sündroomi puhul kõige sagedamini esineva geeni osa deletsioon või puudumine on takistuseks lapse kõndimise ja rääkimise õppimisel. Sellistel lastel on raskem vaimne alaareng ning epilepsiahooge esineb sagedamini ja nende intensiivsus on palju suurem kui teiste kromosomaalanomaaliatega patsientidel.
Kui mutatsioon esineb ainult ühes geenis, saab lapsele piisava tähelepanu ja lähenemise korral õpetada enesehoolduse, suhtlemise ja grupis suhtlemise põhitõdesid, kuigi arengus jääb ta ikkagi oma eakaaslastest maha.
Angelmani sündroomiga laste jaoks, kes on loomult lahked, on kõige olulisem vanemate armastus ja tähelepanu. Ainult sel juhul kannab lapse haridus vilja, isegi kui see on väike. Loomulikult ei saa AS-iga patsiendid tavakoolis õppida. Nad vajavad eriklasse, kus lastele õpetatakse esmalt keskendumist ja seejärel antakse järk-järgult kooliteadmiste põhitõed.
Diagnostika Angelmani sündroom
Angelmani sündroom on kaasasündinud arengupatoloogia. Kuid teatud asjaolude tõttu on seda imikueas ja varases lapsepõlves sageli võimatu diagnoosida. See on tingitud sümptomite mittespetsiifilisusest ja nõrgast väljendusest imikutel ja alla 3-aastastel lastel. Ja haiguse levimus meie riigis ei ole nii suur, et arstid oleksid õppinud seda oma eakaaslaste seas ära tundma.
Angelmani sündroom imikutel võib avalduda lihastoonuse langusena, mis avaldub söötmisprobleemides (imemis- ja neelamisrefleksi nõrkus) ja hiljem raskustes kõndima õppimisel (sellised lapsed hakkavad kõndima palju hiljem). Need sümptomid on esimesed märgid beebi arenguhälbest, mis võib olla seotud kromosomaalse kõrvalekaldega. Ainult geneetiline analüüs saab seda oletust kinnitada.
Erilist tähelepanu pööratakse lastele, kelle vanematel on mitmesuguseid genoomseid või kromosomaalseid häireid. Lõppude lõpuks ei pruugi haigus alguses avalduda ja kui patoloogia avastatakse õigeaegselt, on lapsega intensiivselt töötama hakates võimalik saavutada oluliselt suuremat edu õppimisel, aeglustades haiguse progresseerumist.
Kui vanematel on mitmesuguseid kromosomaalseid kõrvalekaldeid, tehakse geneetiline analüüs juba enne lapse sündi, kuna SA on üks patoloogiatest, mida saab embrüonaalses staadiumis tuvastada.
Geneetilise uurimistöö materjali kogumine võib toimuda kahel viisil:
- invasiivne (teatud riskiprotsendiga, kuna amnionivedeliku proovi võtmiseks on vaja tungida emakasse),
- mitteinvasiivne (lapse DNA analüüs ema verest).
Seejärel viiakse läbi järgmised uuringud:
- fluorestsents in situ hübridisatsioon (FISH-meetod) – spetsiaalse värvainega märgistatud DNA-sondi seondumine uuritava DNA-ga, millele järgneb mikroskoobi all uurimine.
- ube3a geeni ja imprinted-geenide mutatsioonide analüüs,
- DNA metülatsiooni analüüs geneetikas kasutatavate spetsiaalsete meetodite abil.
Geneetilised testid annavad kromosoomanomaaliate korral üsna täpset teavet, mis tähendab, et tulevased vanemad teavad ette, milleks valmistuda. Siiski on ka erandeid. Teatud patsientide rühmal jäävad kõigi patoloogiale viitavate sümptomite esinemisel testi tulemused normaalseks. See tähendab, et patoloogiat saab tuvastada ainult lapse hoolika jälgimise abil juba varasest lapsepõlvest alates: kuidas ta sööb, millal ta hakkas kõndima ja rääkima, kas ta painutab kõndides jalgu jne.
Lisaks FISH-meetodile saab Angelmani sündroomi instrumentaalsete diagnostikameetodite hulgast välja tuua tomograafia (KT või MRI), mis aitab määrata aju seisundit ja suurust, ning elektroentsefalogrammi (EEG), mis näitab aju üksikute osade toimimist.
Tavaliselt teevad arstid lõpliku diagnoosi 3-7-aastaselt, kui patsiendil on juba enamik sümptomeid ja haiguse arengu dünaamika on nähtav.
Millised testid on vajalikud?
Diferentseeritud diagnoos
Angelmani sündroom on geneetiline patoloogia, millel praktiliselt puuduvad spetsiifilised ilmingud. Enamik sümptomeid võib võrdselt viidata nii AS-ile kui ka teistele geneetilistele patoloogiatele.
Angelmani sündroomi diferentsiaaldiagnostika viiakse läbi järgmiste patoloogiatega:
- Pitt-Hopkinsi sündroom (patsiente iseloomustab vaimne alaareng, rõõmsameelne iseloom, naeratus, neil on üsna suur ja lai suu, on täheldatud mikrotsefaaliat). Erinevus seisneb hüperventilatsiooni ja hinge kinnihoidmise rünnakutes ärkvelolekus.
- Christiansoni sündroom (patsiendid on vaimselt alaarenenud inimesed, kellel on rõõmsameelne loom, kes ei suuda rääkida, mida iseloomustab mikrotsefaalia, ataksia, krambid, tahtmatud lihasliigutused).
- Mowat-Wilsoni sündroom (sümptomid: vaimne alaareng, epileptilised krambid, terav lõug, avatud suu, rõõmus ilme näol, mikrotsefaalia). Eristused: suur silmade vaheline kaugus, sissepoole kalduvad silmad, ümar ninaots, tahapoole pööratud kõrvalesta.
- Kabuki sündroom (mida iseloomustab kerge kuni mõõdukas vaimne alaareng, kõne- ja motoorikaprobleemid, lihasnõrkus, epileptilised krambid, mikrotsefaalia, pikad sügeluseintervallid ja koordinatsioonihäired). Iseloomulikud on kaarjad kulmud, alumise silmalau väljapoole pööratud külgmine osa, laia asetusega silmad, pikad silmalau lõhed pikkade ja paksude ripsmetega.
- Retti sündroom (erinevus AS-ist naistel). Sümptomid: kõne arengu hilinemine, krambid, mikrotsefaalia. Erinevus seisneb selles, et näol puudub rõõmus ilme, esinevad uneapnoe ja apraksia hood, mis aja jooksul progresseeruvad.
- Autosomaalselt retsessiivne vaimse alaarengu sündroom 38 (sümptomid: väljendunud vaimne alaareng koos motoorsete oskuste ja kõne arengu hilinemisega, lihasnõrkus, toitumisprobleemid imikueas, impulsiivsus). Eristavaks tunnuseks on iirise sinine värvus.
- MECP 2 geeni duplikatsiooni sündroom (eristumine SA-st meestel). Sümptomid: raske vaimne alaareng, lihasnõrkus lapsepõlvest saati, kõneprobleemid või kõnepuudus, epilepsia. Eristused: progresseeruv müopaatia, pidevalt korduvad infektsioonid.
- Kleefstra sündroom (sümptomid: kõne- ja mõtlemisprobleemid, lihasnõrkus, unehäired, tähelepanu puudumine, avatud suu, hüperaktiivsus, krambid, ataksia, tasakaaluhäired). Iseloomulikud tunnused: lame nägu, lühike töntsnina, laia silmakoopa asetusega silmad, suur väljapoole pööratud alumine huul, agressiivsed ägenemised.
- Smith-Magenise sündroom (mida iseloomustavad krambid, uneprobleemid, intellektuaalse ja motoorse arengu häired). Iseloomulikeks tunnusteks on lai ja lame nägu ning silmatorkav laup.
- Koolen-de-Vries'i sündroom (kerge kuni mõõdukas vaimne alaareng, lihasnõrkus, krambid, sõbralikkus). Iseloomulikud tunnused: piklik nägu kõrge laubaga, punnis kõrvad, kaldus silmad, suur liigeste liikuvus, kaasasündinud südamerikked.
- Phelan-McDermidi sündroom (sümptomid: vaimne alaareng, kõnehäired või kõne puudumine). Erisused: suured käed arenenud lihastega, sünnist saati tekkinud lihasnõrkus, nõrk higistamine.
Sellised patoloogiad nagu adenüülsuktsinaadi puudulikkus, autosomaalselt retsessiivne vaimse alaarengu sündroom 1, kromosoomi 2q23.1 duplikatsiooni sündroom, FOXG1, STXBP1 või MEF2C geeni haploinsufficiency sündroomid ja mõned teised võivad "kiidelda" Angelmani sündroomiga sarnaste sümptomitega.
Arsti ülesanne on teha täpne diagnoos, eristades Angelmani sündroomi sarnaste sümptomitega patoloogiatest ja määrates haiguse diagnoositud staadiumiga seotud tõhusa ravi.
Ravi Angelmani sündroom
Angelmani sündroom on üks neist patoloogiatest, millele meditsiin otsib endiselt tõhusat ravi. Haiguse etioloogiline ravi on arendusjärgus mitmesuguste meetodite ja vahenditega, millest paljusid pole veel inimestel testitud. See tähendab, et praegu peavad arstid piirduma sümptomaatilise raviga, mis aitab kuidagi leevendada marionettsündroomiga laste ja täiskasvanute kadestamisväärset olukorda, kes kannatavad epilepsiahoogude, süljeerituse, hüpotensiooni ja unehäirete all.
Seega on võimalik epilepsiahoogude sagedust ja tugevust vähendada õigesti valitud krambivastase ravimi abil. Kuid kogu raskus seisneb selles, et SA-ga patsientide krambid erinevad tavalistest epilepsiahoogudest selle poolest, et neile on iseloomulik mitut tüüpi krampe, mis tähendab, et seisundit saab leevendada mitme ravimi samaaegse manustamisega.
Angelmani sündroomi raviks kasutatavatest krambivastastest ravimitest on kõige populaarsemad valproehape, topiramaat, lamotrigiin, levetiratsetaam, klonasepaam ja nendel põhinevad ravimid. Harvemini kasutatakse karmasepiinil, fenütoiinil, fenobarbitaalil ja etosuksimiidil põhinevaid ravimeid, kuna mõned neist võivad esile kutsuda paradoksaalse efekti, mis seisneb epilepsiahoogude tugevnemises ja sageduse suurenemises. See juhtub, kui ravimit kasutatakse monoteraapiana.
Süljeerituse raviks kasutatakse tavaliselt kahte meetodit: meditsiinilist (süljeeritust pärssivad ravimid) ja kirurgilist, mis hõlmab süljeeritusjuhade taasimplanteerimist. Kuid SA puhul peetakse neid meetodeid ebaefektiivseteks ja küsimus jääb lahtiseks. Vanemad ja selliste patsientide eest hoolitsejad peavad sellele küsimusele erilist tähelepanu pöörama, kuna patsiendid ise tavaliselt süljeeritust ei kontrolli ja mõned ei suuda lihtsalt enda eest hoolitseda.
Teine probleem on lühike une kestus. Angelmani sündroomiga lapsed magavad sageli mitte rohkem kui 5 tundi, millel on negatiivne mõju kogu keha toimimisele. Kergesti erutuvad, aktiivsed lapsed, kes armastavad mänge ja suhtlemist (isegi kui nad püüavad piirduda mitteverbaalsete meetoditega), on päeva jooksul märgatavalt väsinud. Hea puhkuse saamiseks vajab keha sügavat ja täisväärtuslikku und, aga just see ongi konks.
Näib, et erutunud patsientide une parandamiseks peaksid piisama rahustavatest ravimitest (fenotiasiinid ja atüüpilised antipsühhootikumid), mis rahustavad närvisüsteemi. Kuid AS-i puhul on selliste ravimite kasutamine täis negatiivsete kõrvalmõjude esinemise ohtu. Seetõttu eelistavad arstid siiski leebemaid unerohte, näiteks melatoniini (looduslik hormonaalne ravim, mis põhineb unehormoonil), mida manustatakse patsientidele tund enne magamaminekut 1 tableti kaupa, ja difenhüdramiini. Manustamise sageduse ja annuse määrab arst sõltuvalt patsiendi seisundist ja vanusest.
Mõnikord on Angelmani sündroomiga patsientidel probleeme seedimise ja väljaheitega. Väljaheite parandamiseks võite kasutada lahtisteid (eelistatavalt taimseid).
Või võite probleemile läheneda teisiti, nagu Ameerika arstid tegid, tuginedes mõnele autismi ravimeetodile, sest paljud AS-ile iseloomulikud sümptomid on iseloomulikud ka autismile (impulsiivsus, tahtmatud liigutused, korduvad tegevused, tähelepanuhäired, suhtlemisprobleemid jne). Märgiti, et hormooni sekretiini sissetoomine, mis normaliseerib seedimist ja väljaheidet, mõjutab positiivselt patsientide tähelepanu ning oksütotsiin aitab parandada lapse kognitiivseid võimeid ja mälu ning korrigeerida käitumist.
Tõsi, ainult hormoonidest ei piisa, eriti laste puhul. Angelmani sündroomi korral on näidustatud käitumisteraapia, töö psühholoogi ja logopeediga (mitteverbaalse suhtluse meetodite ja viipekeele õpetamine). Selliste laste haridus peaks põhinema individuaalsel programmil, milles osalevad spetsiaalselt koolitatud õpetajad, psühholoog ja vanemad. Kahjuks pole see kõikjal võimalik ja pered jäävad oma probleemiga üksi.
Kuna paljudel noortel AS-i patsientidel on madal lihastoonus ja liigeseprobleemid, pööratakse suurt tähelepanu füsioteraapiale. Kõige sagedamini kasutavad arstid parafiiniaplikatsioone, elektroforeesi ja magnetravi.
Aktiivne toonikmassaaž ja terapeutilise füüsilise ettevalmistuse spetsiaalsed harjutused aitavad haigel lapsel mõne aja pärast jalgadele tõusta ja enesekindlalt kõndida. Eriti kasulik on selles osas vesivõimlemine, mida soovitatakse jahedas vees SA korral. See suurendab lihastoonust ja õpetab last oma keha kontrollima ning liigutusi koordineerima.
Krambivastane ravi
Angelmani sündroomi kõige ohtlikum sümptom on epilepsiaga sarnased krambid. Seda sümptomit täheldatakse 80% patsientidest, mis tähendab, et kõigile neile tuleb määrata efektiivne krambivastane ravi.
Epilepsiahoogude ravi toimub vitamiinide ja krambivastaste ravimite abil. Angelmani sündroomi korral, millega kaasneb krampisündroom, on kasulikud B-vitamiinid, samuti C-, D- ja E-vitamiinid. Kuid vitamiiniravi iseseisev määramine on sel juhul väga ohtlik, sest kontrollimatu vitamiinide tarbimine võib vähendada epilepsiavastaste ravimite efektiivsust ja provotseerida uusi, raskemaid ja pikemaajalisi krampe.
Krambivastaste ravimite valiku ja nende efektiivse annuse määramise peaks samuti tegema eriarst. Samuti otsustab ta, kas piisab ühest ravimist või peab patsient pikka aega võtma kahte või enamat ravimit.
Enamiku patsientide jaoks määravad arstid valproehappe ravimeid (Valproehape, Depakine, Convulex, Valparin jne), mis ennetavad krampe ning parandavad patsientide meeleolu ja vaimset seisundit.
Valproehape on saadaval tablettide, siirupi ja süstelahuste kujul. Kõige populaarsem ravim on pikatoimeline ravim "Depakine" tablettidena ja intravenoosseks manustamiseks mõeldud lahusena. Ravimi annuse määrab arst individuaalselt, olenevalt patsiendi kehakaalust, vanusest ja seisundist.
Ravimit võetakse söögi ajal 2–3 korda päevas. Keskmine päevane annus on 20–30 mg patsiendi kehakaalu kilogrammi kohta, maksimaalne on 50 mg/kg päevas.
Kasutamise vastunäidustused. Mitte kasutada maksa- ja kõhunäärme talitlushäirete, hemorraagilise diateesi, hepatiidi, porfüüria ja ravimi suhtes ülitundlikkuse korral.
Kõrvaltoimete hulka kuuluvad käte värisemine, seede- ja väljaheitehäired ning kehakaalu muutused.
"Topiramaat" on ka SA jaoks valitud ravim. Seda toodetakse tableti kujul ja seda kasutatakse nii monoteraapiana kui ka kombinatsioonis teiste ravimitega.
Manustamisviis ja annustamine. Tablette tuleb võtta suu kaudu, olenemata toidukordadest. Täiskasvanute algannus on 25–50 mg, laste puhul 0,5–1 mg/kg. Igal nädalal suurendatakse annust vastavalt arsti juhistele.
Ravimit ei tohi võtta raseduse ja imetamise ajal, samuti ülitundlikkuse korral selle komponentide suhtes.Ravimil on palju erinevaid kõrvaltoimeid.
Ravimid, mida arst võib Angelmani sündroomi korral välja kirjutada: klomazepam, rivotril, lamotrigiin, seizar, lamictal, levetiratsetaam, keppra, epiterra jne.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditsiooniline meditsiin ja homöopaatia
Traditsiooniline meditsiin, nagu homöopaatilised preparaadid, on muidugi suhteliselt ohutu, kuid sellise ravi efektiivsust Angelmani sündroomi puhul võib pidada vastuoluliseks.
Kuigi rahvapärane ravi võib mõnes asjas siiski aidata. Me räägime epilepsiahoogude peatamisest. Selles osas võib taimne ravi olla üsna tõhus.
Hea efekti annab pojengil, lagritsal ja leedel põhinev ravimkollektsioon (komponendid võetakse võrdsetes kogustes). Ürdid tuleb jahvatada jahuks. 2 nädala pärast selle võtmise algusest võite märgata krampide sageduse olulist vähenemist.
Krampide korral on kasulik ka lavendli keetmine (1 teelusikatäis klaasi keeva vee kohta). Segu keedetakse 5 minutit ja lastakse pool tundi tõmmata. Ravimit võetakse öösel 14 päeva jooksul.
Emajõe vesilahust (või alkoholilahust) peetakse epilepsiahoogude korral tõhusaks.
Angelmani sündroomi krampide ennetamiseks mõeldud homöopaatilistest preparaatidest võib kasutada kummeli ja emajuure, hüdrotsüanikumi, nitriidi, kaltsiumi ja arseeni baasil valmistatud ravimeid. Kuid tuleb arvestada, et ainult homöopaatiline arst saab igal konkreetsel juhul määrata ravimite tõhusaid ja ohutuid annuseid.
Ärahoidmine
Nagu lugeja ilmselt juba aru sai, ei ole meditsiin veel võimeline geenimutatsioone ja muid kromosomaalseid kõrvalekaldeid ennetama ning olukorda parandama. See võib juhtuda igaühega, sest Angelmani sündroomiga lapsed sünnivad tervetele vanematele ja geneetika, mis on praegu üks vähem uuritud meditsiiniharusid, ei oska seda veel seletada.
Ainus, mida teha saab, on suhtuda raseduse planeerimisse vastutustundlikult, registreeruda ja õigeaegselt läbida uuringud. Kuid jällegi on selline meede pigem hariv kui ennetav, nagu iga teine uuring. Kuid noored vanemad teavad ette, milleks valmistuda, ja positiivse vastuse korral otsustavad nad, kas nad saavad võtta sellise vastutuse nagu haige lapse kasvatamine.
Prognoos
Angelmani sündroomi prognoos sõltub kromosomaalse anomaalia olemusest ja selle avastamise õigeaegsusest. Kõige raskemini kannatavad need lapsed, kelle 15. kromosoomis on geenides "lüngad" (deletsioon). Selliste patsientide kõndimise ja rääkimise tõenäosus on äärmiselt väike. Teisi juhtumeid saab korrigeerida hoolika lähenemise ja lapse armastusega.
Kahjuks ei ole sellistel patsientidel võimalik saada täisväärtuslikeks ühiskonnaliikmeteks, hoolimata asjaolust, et nad pole kaugeltki rumalad, nad saavad kõnest ja selle tähendusest aru. Suhtlemisprobleemid jäävad neile aga kogu eluks. Patsientidele saab viipekeelt õpetada juba lapsepõlvest saati, kuid neid ei saa sundida sõnade abil suhtlema. "Rääkivate" patsientide sõnavara piirdub igapäevaelus kasutatavate sõnade miinimumiga (5-15 sõna).
Angelmani sündroomiga patsientide oodatava eluea ja üldise tervise osas kõiguvad need näitajad keskmiste väärtuste ümber. Täiskasvanueas seisavad patsiendid enamasti silmitsi selliste terviseprobleemidega nagu skolioos ja rasvumine, mis õige ravi korral ei ole eluohtlikud.