Artikli meditsiiniline ekspert

Uued väljaanded
Treacher Collinsi sündroom
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.

Luu arenguprotsesside emakasisesed häired põhjustavad tõsiseid kraniofakiaalseid deformatsioone ja üks sellise patoloogia sortidest on Treacher Collinsi sündroom (TCS) või mandibulofascial, st maxillofaciaalne düsostoos.
Haiguskood vastavalt RHK 10-le: XVII klass (kaasasündinud anomaaliad, deformatsioonid ja kromosomaalsed häired), Q75.4 - mandibulofatsiaalne düsostoos.
Põhjused Treacher Collinsi sündroom
See sündroom sai nime silmapaistva Briti silmaarsti Edward Treacher Collinsi järgi, kes kirjeldas patoloogia põhitunnuseid enam kui sada aastat tagasi. Euroopa arstid nimetavad seda tüüpi näo- ja lõualuu luude anomaaliat aga sagedamini Franceschetti haiguseks või sündroomiks – see põhineb Šveitsi silmaarsti Adolf Franceschetti ulatuslikel uuringutel, kes võttis eelmise sajandi keskel kasutusele termini "mandibulofastsiaalne düsostoos". Meditsiiniringkondades kasutatakse ka nimetust Franceschetti-Collinsi sündroom.
Treacher Collinsi sündroomi põhjustavad mutatsioonid TCOF1 geenis (5q31.3-33.3 kromosoomi lookuses), mis kodeerib nukleoolset fosfoproteiini, mis vastutab inimese embrüo kraniofakiaalse osa moodustumise eest. Selle valgu hulga enneaegse vähenemise tagajärjel on häiritud rRNA biogenees ja funktsioonid. Inimese Genoomi uurimisprogrammi geneetikute sõnul viivad need protsessid närviharja embrüonaalsete rakkude proliferatsiooni vähenemiseni - see on seljandik mööda närvivagu, mis embrüonaalse arengu ajal sulgub närvitoruks.
Näokudede moodustumine toimub närviharja ülemise (pea) osa rakkude transformatsiooni ja diferentseerumise tõttu, mis migreeruvad mööda närvitoru embrüo esimese ja teise harukaare piirkonda. Nende rakkude puudus põhjustab kraniofakiaalseid deformatsioone. Anomaaliate tekke kriitiline periood on 18 kuni 28 päeva pärast viljastumist. Pärast närviharja rakkude migratsiooni lõppemist (neljandal rasedusnädalal) moodustuvad peaaegu kõik näopiirkonna lahtised mesenhümaalsed koed, mis hiljem (5–8 nädalal) diferentseeruvad näo, kaela, kõri, kõrva (sh sisekõrva) ja tulevaste hammaste skeleti- ja sidekudedeks.
Pathogenesis
Treacher Collinsi sündroomi patogenees on sageli perekondlik ja anomaalia pärandub autosomaalselt dominantsel viisil, kuigi esineb ka defekti autosomaalselt retsessiivset ülekannet (mutatsioonidega teistes geenides, eriti POLR1C ja POLR1D). Kõige ettearvamatum asi lõualuu düsostoosi puhul on see, et mutatsioon pärandub lastel ainult 40–48% juhtudest. See tähendab, et 52–60% patsientidest ei ole Treacher Collinsi sündroomi põhjused seotud anomaalia esinemisega perekonnas ja arvatakse, et patoloogia tekib juhuslike geenimutatsioonide tagajärjel de novo. Tõenäoliselt on uued mutatsioonid lootele raseduse ajal avaldunud teratogeense toime tagajärjed.
Selle sündroomi teratogeensete põhjuste hulgas nimetavad eksperdid suuri etanooli (etüülalkoholi) annuseid, kiirgust, sigaretisuitsu, tsütomegaviiruse ja toksoplasma, samuti glüfosaadil põhinevaid herbitsiide (Roundal, Glyfor, Tornado jne). Ja iatrogeensete tegurite loetelu hõlmab akne ja seborröa ravimeid 13-cis-retinoehappega (isotretinoiin, Accutane); krambivastast ravimit fenütoiini (Dilantin, Epanutin); psühhotroopseid ravimeid diazepam, valium, relanium, seduksen.
Sümptomid Treacher Collinsi sündroom
Enamasti sõltuvad mandibulofastsiaalse düsostoosi kliinilised tunnused ja nende avaldumisaste geenimutatsioonide avaldumise iseärasustest. Ja selle anomaalia esimesed märgid on enamasti lapsel nähtavad kohe pärast sündi: Treacher Collinsi sündroomiga näol on iseloomulik välimus. Lisaks on morfoloogilised anomaaliad tavaliselt kahepoolsed ja sümmeetrilised.
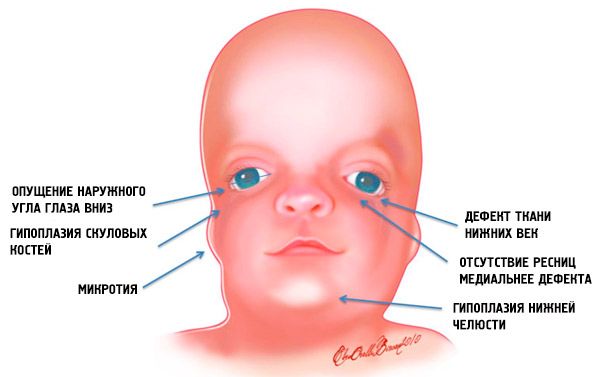
Treacher Collinsi sündroomi kõige ilmsemad sümptomid on:
- kolju näo luude alaareng (hüpoplaasia): eesmise luu sarnaluu, sarnaluujätked, külgmised pterygoidplaadid, paranasaalsed siinused, alumine lõualuu ja luu epifüüside (kondüülide) eendid;
- alalõualuu luude vähearenenud areng (mikrognaatia) ja tavapärasest nürim alalõua nurk;
- nina on normaalse suurusega, kuid tundub suur kulmukaare hüpoplaasia ja oimuspiirkonna sarnakaarte vähearenenud või puudumise tõttu;
- silmapilud on allapoole suunatud, see tähendab, et silmade kuju on ebanormaalne, välisnurgad on allapoole rippuvad;
- alumiste silmalaugude defektid (koloboom) ja ripsmete osaline puudumine neil;
- ebakorrapärase kujuga kõrvakesed, millel on lai valik kõrvalekaldeid, sealhulgas nende asukoht alalõualuu nurgas, lobede puudumine, pimedad fistulid kõrva traguse ja suu nurga vahel jne;
- välise kuulmekäigu ahenemine või sulgumine (atresia) ja keskkõrva kuulmekäikude anomaaliad;
- kõrvasüljenäärmete puudumine või hüpoplaasia;
- neelu hüpoplaasia (neelu ja hingamisteede ahenemine);
- kõva suulae mitteühendumine (suulaelõhe), samuti pehme suulae puudumine, lühenemine või liikumatus.
Sellistel anatoomilistel anomaaliatel on alati tüsistusi. Need on funktsionaalsed kuulmishäired juhtiva kuulmislanguse või täieliku kurtuse näol; nägemishäired silmamunade ebaõige moodustumise tõttu; suulae defektid, mis põhjustavad raskusi söömise ja neelamisega. Lõualuudefektidega kaasnevad hambumushäired (maloklusioon), mis omakorda põhjustavad probleeme närimise ja artikulatsiooniga. Pehme suulae patoloogiad selgitavad nasaalset häält.
Tüsistused ja tagajärjed
Treacher Collinsi sündroomi maxillofacial anomaaliate tagajärjed on see, et lapse sündides on intellektuaalsed võimed normaalsed, kuid kuulmisdefektide ja muude häirete tõttu täheldatakse sekundaarset vaimset alaarengut.
Lisaks tunnevad selliste defektidega lapsed teravalt oma alaväärsust ja kannatavad, mis mõjutab negatiivselt nende närvisüsteemi ja psüühikat.
Diagnostika Treacher Collinsi sündroom
Treacher Collinsi sündroomi postnataalne diagnoosimine põhineb sisuliselt kliinilistel tunnustel. Kraniofakiaalne düsostoos on kergesti tuvastatav, kui sündroom on täielikult väljendunud, kuid kui esinevad minimaalselt väljendunud patoloogilised sümptomid, võib tekkida probleeme õige diagnoosi seadmisega.
Sellisel juhul tuleks pöörata erilist tähelepanu kõigi anomaaliatega seotud funktsioonide hindamisele, eriti nendele, mis mõjutavad hingamist (uneapnoe ohu tõttu). Samuti tuleks hinnata ja jälgida toitmise efektiivsust ja hemoglobiini hapnikuküllastust.
Hiljem, 5.-6. päeval pärast sündi, tuleb kuulmiskahjustuse ulatus kindlaks määrata audioloogilise testi abil, mis tuleks läbi viia sünnitusmajas.
Määratakse uuring, mille käigus teostatakse instrumentaalne diagnostika kraniofakiaalse düsmorfoloogia fluoroskoopia abil; pantomograafia (näo kolju luustruktuuride panoraamröntgen); täielik kolju kompuutertomograafia erinevates projektsioonides; aju KT või MRI sisemise kuulmekäigu seisundi määramiseks.
Lõualuu anomaaliate varaseim – sünnieelne – diagnoosimine perekonnaanamneesis Treacher Collinsi sündroomi korral on võimalik koorionivilluse biopsiaga 10–11 rasedusnädalal (protseduur ähvardab raseduse katkemist ja emaka infektsiooni).
Vereanalüüse võetakse ka pereliikmetelt; 16.–17. rasedusnädalal analüüsitakse lootevett (transabdominaalne amniotsentees); 18.–20. rasedusnädalal tehakse fetoskoopia ja võetakse verd platsenta loote veresoontest.
Kuid kõige sagedamini kasutatakse ultraheli selle sündroomi sünnieelseks diagnoosimiseks lootel (rasedusnädalal 20–24).
Millised testid on vajalikud?
Diferentseeritud diagnoos
Samu meetodeid kasutavad spetsialistid ka siis, kui on vaja diferentsiaaldiagnostikat kerge Treacher Collinsi sündroomi äratundmiseks ja eristamiseks teistest kraniofakiaalsete luude kaasasündinud anomaaliatest, eelkõige: Aperti, Crouzoni, Nageri, Petersi-Hewelsi, Hellermanni-Stephi sündroomidest, samuti hemifakiaalsest mikrosoomiast (Goldenhari sündroom), hüpertelorismist, kraniaalsete õmbluste enneaegsest sulandumisest (kraniosünostoos) või näoluude sulandumise häiretest (kraniosünostoos).
Ravi Treacher Collinsi sündroom
Nagu kõigil geneetiliselt määratud kaasasündinud defektide juhtudel, on ka Treacher Collinsi sündroomi raskete vormide ravi eranditult palliatiivne, kuna selliste patoloogiate raviks lihtsalt puuduvad meetodid. Selle sündroomi deformatsioonide spekter ja aste on ulatuslikud ning seetõttu on ka meditsiinilise sekkumise olemus ja intensiivsus palju valikuvõimalusi.
Kuulmise korrigeerimiseks ja parandamiseks kasutatakse kuuldeaparaate ning kõne parandamiseks logopeedilisi seansse.
Kirurgilised sekkumised on varases eas vajalikud hingamisteede (teostatakse trahheostoomia) ja kõri (toitmiseks tehakse gastrostooomia) ahenemise rasketel juhtudel. Samuti võib osutuda vajalikuks suulae kirurgiline korrigeerimine.
Alalõua pikendamise operatsioone tehakse 2-3-aastaselt või hiljem. Pehmete kudede rekonstrueerimine hõlmab alumise silmalau koloboomi korrigeerimist ja kõrvaplastikat.
Prognoos
Milline on selle patoloogia prognoos? See sõltub deformatsiooni astmest ja sümptomite intensiivsusest. Treacher Collinsi sündroom on eluaegne diagnoos.
 [ 25 ]
[ 25 ]