Artikli meditsiiniline ekspert

Uued väljaanded
Xeroderma pigmentosum
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
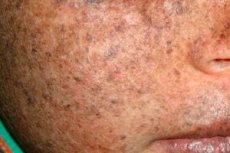
Xeroderma pigmentosum on autosoom-retsessiivne DNA parandamise geneetiline häire. Haiguse põhjustavad mutatsioonid geenides, mis on seotud ultraviolettkiirguse ja muude toksiliste ainete poolt kahjustatud DNA parandamisega.
Kõige sagedamini mõjutab see haigus lapsi, keda nimetatakse ka "öö lasteks". Haiguse sagedased tüsistused on basaliom ja muud naha pahaloomulised kasvajad, metastaatiline pahaloomuline melanoom ja lamerakk-kartsinoom.
 [ 1 ]
[ 1 ]
Põhjused xeroderma pigmentosum
Pathogenesis
Haiguse tekkes mängib olulist rolli UV-endonukleaasi ensüümide puudulikkus patsiendi rakkudes (fibroblastides) või nende täielik puudumine. See ensüüm vastutab ultraviolettkiirte poolt kahjustatud DNA paljunemise eest. Teiste allikate kohaselt mängib mõnedel patsientidel haiguse tekkes olulist rolli DNA polümeraas-1 ensüümi puudulikkus. Haigus areneb kõige sagedamini 280-310 nm lainepikkusega kiirte mõjul. Arvatakse, et pigmentkseroderma tekib mitmesuguste fotodünaamiliste ainete sattumise tõttu organismi või porfüriinide hulga suurenemise tõttu inimese bioloogilises keskkonnas.
Haiguse algstaadiumis täheldatakse hüperkeratoosi, epidermise fookuse atroofiat, Malpighi kihi hõrenemist, melaniini graanulite suurenemist rakkude basaalkihis ja kroonilist põletikulist infiltraati dermise ülemises kihis (peamiselt veresoonte ümbruses). Seejärel avastatakse kollageeni- ja elastsetes kiududes degeneratiivseid muutusi ning kasvaja staadiumis ilmnevad nahavähile iseloomulikud tunnused.
Sümptomid xeroderma pigmentosum
See haigus mõjutab nii mehi kui ka naisi võrdselt. Haigus algab varases lapsepõlves kevadel või suvel. Patsiendi nahk on päikesevalguse suhtes eriti tundlik. Seetõttu ilmub esimene lööve hüperpigmentatsiooni kujul, mis sarnaneb sünnimärgiga, päikesevalgusele avatud nahapiirkondadele. Lööve suureneb, erüteem süveneb, muutub tumepruuniks, tekivad telangiektaasiad, angioomid ja atroofia. Seejärel kasvavad papilloomid ja tüükad (peamiselt näo ja kaela nahal), tekivad laigud ja haavandid. Haavandite armistumise tagajärjel muutub nina õhemaks ("linnunokk"), silmalaug paindub. Papilloomid arenevad tavaliselt pahaloomulisteks kasvajateks. Pigmenteerunud kseroderma esineb reeglina koos spinotsellulaarse vähi, melanosarkoomidega.
Mis teid häirib?
Etapid
Xeroderma pigmentosumi kliinilises kulgemises eristatakse viit etappi: põletikuline (erüteemiline), hüperpigmentatsioon, atroofia, hüperkeratoos ja pahaloomulised kasvajad.
Põletikulise (erüteemilise) staadiumi ajal tekivad päikesevalgusega kokkupuutunud nahapiirkondadele (nägu, kael, rindkere ülaosa, käed, käelabad) turse, punased laigud ning mõnikord ka villid ja vesiikulid. Päikesevalgusega kokku puutumata piirkondades löövet peaaegu ei esine.
Hüperpigmentatsiooni korral ilmuvad punaste laikude asemele helepruunid, pruunid hüperpigmenteerunud laigud, mis sarnanevad sünnimärkidega.
Atroofia korral nahk kuivab, muutub õhemaks ja kortsub. Huulte ja nina nahal täheldatakse arvukalt väikeseid tähtkujulisi telangiektaasiaid ja läikiva pinnaga arme. Atroofia ja armide tõttu tekivad mikrotoomia (suuava ahenemine), ektropioon, kõrvade ja nina hõrenemine, ninaava ja suu atresia. 80–85% patsientidest on mõjutatud silmad: täheldatakse konjunktiviiti, keratiiti, sarvkesta ja limaskesta kahjustusi ning nägemise nõrgenemist. Silmalaugude nahal täheldatakse düskroomiat, telangiektaasiat, hüperkeratootilisi muutusi ja kasvajaid.
Hüperkeratoosi korral ilmuvad ülalkirjeldatud patoloogilistesse fookustesse tüükakasvajad, papilloom, keratoakantoom, fibroom, angiofibrioom ja muud healoomulised kasvajad. Seetõttu kuuluvad mõned teadlased pigmendi kseroderma eelvähihaiguste rühma.
10-15 aastat pärast haiguse algust ilmuvad atroofiliselt muutunud kolletesse ja pigmendilaikudesse pahaloomulised nahakasvajad (basaloom, endotelioom, angiosarkoom). Kasvajad hävivad lühikese aja jooksul, metastaseeruvad siseorganitesse ja viivad surmani. Mõnedel patsientidel täheldatakse siseorganites ja kudedes üldiseid düstroofilisi muutusi (teise ja kolmanda varba sündakteelia, hambadüstroofia, täielik juuste väljalangemine jne).
Vormid
Xeroderma pigmentosumi neuroloogiline vorm avaldub kahes sündroomis.
Reedi sündroomi iseloomustavad kseroderma pigmentosumi ja mikrotsefaalia kliinilised ilmingud, idiopaatia ja skeletisüsteemi aeglane kasv. Haiguse neuroloogilises vormis on DNA reparatsioon keeruline ja röntgenteraapia viib peamiste patoloogiliste fookuste suurenemiseni.
De Sinctis-X Cocchione sündroomi iseloomustavad järgmised kliinilised tunnused:
- kseroderma pigmentosumi kliiniliste tunnuste teke eriti valgustundlikul nahal;
- pahaloomuliste kasvajate varajane ilmumine;
- spastilise paralüüsi, mikrotsefaalia ja dementsuse samaaegne kulg;
- kaasasündinud väärarengud ja kääbuskasvud;
- sugunäärmete vähearenenud areng;
- sagedased raseduse katkemised;
- retsessiivne edasikandumine pärimise teel.
Mõnede dermatoloogide sõnul ei ole Santis-Cacchione sündroom iseseisev haigus, vaid kseroderma pigmentosumi raske ja täielikult väljendunud kliiniline ilming.
Geneetilised vormid
Tüübid |
Geen |
Lookus |
Kirjeldus |
Tüüp A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) grupp A – klassikaline vorm |
Tüüp B, II, XPB |
XPB |
2q21 |
XP B-grupp |
Tüüp C, III, XPC |
XPC |
3p25 |
XP C-grupp |
Tüüp D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP rühm D või De Sanctis-Cacchione sündroom |
Tüüp E, V, XPE |
DDB2 |
11p12-p11 |
XP E-grupp |
Tüüp F, VI, XPF |
ERCC4 |
16 lk 13.3–13.13 |
XP F-grupp |
Tüüp G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP G-grupi ja COFS-i sündroomi (tserebro-okulo-fatsioskeletaalse sündroomi) tüüp 3 |
Tüüp V, XPV |
POLH |
6p21.1-p12 |
Xeroderma Pigmentosa variant |
Mida tuleb uurida?
Kuidas uurida?
Kellega ühendust võtta?
Ravi xeroderma pigmentosum
Malaariavastased ravimid (delagüül, plakeniil, resokviin jne), mis kaitsevad DNA-d ultraviolettkiirte eest, pärsivad depolümerisatsiooniprotsessi, vähendavad naha tundlikkust (fotosensibiliseerumist) päikesevalguse suhtes. Nendel ravimitel on põletikuvastased ja hüposensibiliseerivad omadused. Soovitatav on läbi viia üldravi koos vitamiinraviga (B1, B2, PP, B6, B12, A, E), antihistamiinikumidega (tavegil, difenhüdramiin, suprastiin), desensibiliseerivate ainetega (naatriumtiosulfaat, 10% kaltsiumkloriidi intravenoosselt 10 ml).
Kohalikuks raviks kasutatakse päikesekaitsekreemi ja -salve.
Pigment-kseroderma kasvajavormis kasutatakse kirurgilisi meetodeid, vedelat lämmastikku ja laserkiiri. Päikesevalguse eest kaitsmiseks peate kandma lahtisi riideid, päikesemütse ja kindaid.
Prognoos
Enamik patsiente (2/3) sureb enne 15. eluaastat. Mõned patsiendid võivad elada 40–50-aastaseks.
[ 20 ]