
Angelmanni sündroom lastel ja täiskasvanutel
Viimati vaadatud: 23.04.2024

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.

On mitmeid haigusi, mille puhul väljendid nagu "enda eest hoolitsemine ja mitte haige" on vähemalt naeruväärsed. See patoloogia, mille puhul mõni vaimne ja füüsiline kõrvalekalle on lapse kehasse varemgi enne sünnitust, kuid vanemad ei ole süüdi. Sellised haigused on põhjustatud kromosoomide komplektides esinevatest mutatsioonidest või häiretest ja neid nimetatakse kromosoomideks või geneetilisteks. Angelmani sündroom, Downi sündroom, Patau, Edwards, Turner, Prader-Willi on geneetiliste haiguste osa vaid üsna korralikust nimekirjast.
Õnneliku inimese sündroom
Seekord räägime haiguse nime Briti lastearst Harry Angelmani sellest esimesest tõstatas probleemi 1965 aastal silmitsi eelõhtul tema praktikas kolme ebatavaline lapsi, keda ühendab ühine omapärane sümptomid. Arst nimetas need laste nukulased ja kirjutas neile artikli, mida algselt nimetati nukutele. Artikkel ise ja selle nimi olid kirjutatud Verona ühes muuseumis nägematuna. Pilt kujutas naeruvat poissi ja seda kutsuti "Poiss-nuku". Pildil kujutatud lapse seos kolme lapsega, keda Angelman kordas oma praktikas kokku, kutsus lapsehoidjat üles ühendama lapsi ühe rühma tõttu olemasoleva haiguse tõttu.
See, et artiklis märgitud lapsed ei märganud teisi arste, ei ole üllatav. Lõppude lõpuks tundus esmapilgul, et neil olid täiesti erinevad haigused, seega haiguse üldine kliiniline pilt erines kolmel erineval juhul. "Uus" kromosomaalne patoloogia võib olla huvitav teistele teadlastele, kuid sel ajal ei olnud geneetika veel piisavalt välja töötatud, et kinnitada inglise arsti hüpoteesi. Seepärast jäeti artikkel pärast teatud huvi pikka aega mahajäetud rügemendi juurde.
Angelmani sündroomi järgmine märkus, mida nüüd nimetati Inglismaa pediaatriartikliteks G. Anglemanniks, pärineb 20. Sajandi 80. Aastate algusest. Ja ainult 1987. Aastal oli võimalik leida põhjus, miks väikeste lastega on selliseid kõrvalekaldeid sündinud, et küljelt näivad nad pidevalt naeratust ja õnnelikku. Tegelikult pole see nii, ja naeratus on lihtsalt grimats, mille taga on õnnetu inimeste hing ja vanemate valu.
Epidemioloogia
Vastavalt statistikale võib laps kromosomaalne mutatsioon areneda nii vanemate selliste mutatsioonide taustal kui ka selliste puudumisel. Angelmani sündroomi (SA) ei ole selget pärilikku iseloomu, kuid kroonilise mutatsiooniga vanemate patoloogia arenguprognoos on üsna kõrge.
Samuti on huvitav, et kui perekonnal on juba SA-ga laps, on üheprotsendiline võimalus omada sama tüüpi teist last, isegi kui vanemad on terved.
Anghelmani sündroomiga patsientide arvu kohta pole täpseid statistilisi andmeid. Võibolla on süüdi mitmesuguseid sümptomeid, mis võivad tekkida teatavas kompositsioonis või pikka aega üldse mitte tekkida. Eeldatakse, et haiguse levimus on: 1 laps 20 000 vastsündinu kohta. Kuid see arv on väga ligikaudne.
Põhjused angelmaani sündroom
Angelmanni sündroom on kromosomaalse patoloogia meditsiiniline nimi, kuid see ei ole ainus. Inimestel on seda haigust kutsutud ka nukute laste sündroomiks, õnneliku nukkide sündroomiks ja Petrushka sündroomiks ja naeruvähkli sündroomiks. Jah, millised nimed inimesed ei saa välja tulla (mõnikord isegi solvav patsiendi endi ja tema vanemate jaoks), kuid haigus on haigus, ükskõik kui naljakas see välja näeb välja ja mis põhjustel võib olla põhjustatud.
Angelmani sündroomi arengu põhjused ning paljud teised geneetilised patoloogiad on alati üksiku kromosoomi või kogu kromosoomi struktuuri rikkumine. Kuid ainult meie juhul on kogu probleem ema ülekantud 15 kromosoomist. Ie. Sellisel juhul pole isamaalse kromosoomi kõrvalekaldeid, kuid naisel on teatud mutatsioonid.
Vastavalt kromosomaalsete kõrvalekallete tüübile viitab Angelmanni sündroom kromosomaalsetele mutatsioonidele. Sellised mutatsioonid on:
- Kustutamine (puudumine kromosoomi piirkond, mis sisaldab konkreetsete geenide ja kui keegi geenide tegemist mikrodeletsioonid), mis on tingitud kahe katkestused ja üks taasühinemise kui kaotas osa originaal kromosoomi.
- Paljundamine (kromosoomis täiendava saidi olemasolu, mis on juba olemasoleva koopia), mis enamasti viib inimese surmani, harvem - viljatuseni.
- Inversioon (inversioon kromosoomi lõigud temperatuuril 180 kraadi, st vastupidises suunas ja seejärel geenide sellesse paigutatud vastupidises järjekorras) kui katkine kromosoomi otsad ühendatakse erinevas järjekorras originaal.
- Sisestamine (kui osa kromosoomi geneetilisest materjalist ei ole selle asemel),
- translokatsioon (kui osa kromosoomi liitub teise kromosoomi, võib selline mutatsioon olla vastastikune, ilma saite kadumiseta).
Muutuva kromosoomi saamine pahaaimamatust emalt, lapsepõlves hukkamõistetud kõrvalekalded on varem sündinud. Anghelmani sündroomi arengu kõige levinum põhjus on endiselt emade 15 kromosoomi kustutamine, kui selles ei ole väikest ala. "Naerulise nuku" sündroomi levinuimad mutatsioonid on:
- translokatsioon
- üksikvanema diosoomia (kui laps sai paar kromosoome isast, puudub ema kromosoom)
- DNA geenide mutatsioon, mis on nii peamine ehitusmaterjal (geneetiline) materjal kui ka õige kasutamise juhend (eelkõige ube3a geeni mutatsioon emade kromosoomis).
Ühe sellise mutatsiooni esinemine vanemates on Anghelmani sündroomi riskitegur lastel. Kuid mitte ainult kromosomaalsed mutatsioonid, vaid ka genoomsed mutatsioonid (mis on seotud kvantitatiivse muutusega kromosoomide komplektides ja leitakse sagedamini kui kromosoomid) võivad põhjustada lapse haiguse arengut. Tavalistele genoomsetele mutatsioonidele võib seostada kromosoomide trisoomia (kui inimese kromosoomikompleksil on üle 46 kromosoomi).
Lapse patoloogias ei pruugi vanematel olla kromosoomide kõrvalekaldeid. Siiski on teatud protsent patsientidest, kelle haigus on pärilik.

Pathogenesis
Läheme veidi bioloogiasse, täpsemalt geneetikasse. Iga inimese keha geneetiline teave sisaldub 23 paari kromosoomis. Üks paarist pärinev kromosoom edastatakse lapsele isalt, teine emalt. Kõik kromosoomide paarid erinevad kuju ja suuruse poolest ja kannavad ise teatud teavet. Niisiis, vastutab teket lapse soo tunnused 23 paari kromosoome (X ja Y kromosoomid) (XX - tüdruk, poiss HY-, Y-kromosoomi, beebi saavad ainult isalt).
Ideaaljuhul saab laps oma vanematest 46 kromosoomi, mis moodustavad tema geneetilise atribuudi ja määrab tema individuaalsena. Suurem arv kromosoome nimetatakse trisoomiks ja loetakse kõrvalekalleks normist. Näiteks põhjustab Downi sündroomi tekkimist 47 kromosoomi olemasolu kromosoomide komplekti (karüotüüp, mis määrab kindlaks liigi ja individuaalsed omadused).
Kui kromosoome toonitakse spetsiaalse värviga, siis saab mikroskoobist näha igaüks neist erinevates toonides. Igas grupis on suur hulk geene. Kõik need lindid on nummerdatud teadlaste poolt ja neil on kindel asukoht. Ühe sagedusribade puudumist peetakse standardi kõrvalekalleks. Angelmanni sündroomiga võite väga tihti jälgida emase kromosoomi segmentide puudumist intervallis q11-q13, mis asub pika käe all, kusjuures DNA-aluste arv on vaid umbes 4 miljonit.
Kromosoomi peamine komponent on uskumatult pikk DNA molekul, mis sisaldab tuhandeid geene ja kümneid ja sadu miljoneid lämmastikkuvaid aluseid. Seega on Angelmani sündroomi ja mitmete teiste 15 kromosoomide puhul 1200 geeni ja umbes 100 miljonit alust. Kõik DNA molekuli struktuuri rikkumised mõjutavad tingimata sündimata lapse välimust ja arengut.
Geenis sisalduv geneetiline teave muundatakse valku või RNA-ks. Seda protsessi nimetatakse geeniekspressiooniks. Seega vanematest saadud geneetiline teave saab nii vormis kui ka sisusena, mis on väljendatud naiste või meeste suguelundite ainulaadses pärijais.
On mitmeid patoloogiate koos mitteklassikaline tüüpi pärilikkuse, sealhulgas Angelmani sündroom, kus geenid saadud vanemate osana paarilisest kromosoomid unikaalse jäljendi vanemad ja avalduvad erinevalt.
Niisiis, Angelmani sündroom on kõige ilmekam näide genoomse jäljendamisaken, kusjuures geenide ekspressiooni kehas laps otseselt sõltuvalt kellelt vanema tuletatud alleelide (eri vormides sama geeni saadud isast ja emast asuvad identsed portsjoni paaris kromosoomid) . Ie. Põhjustada tekkimist sündroom häiretena emade kromosoomi, samas mutatsioonid ja häirete paternal kromosoomi struktuuri põhjuseks väga erinevaid haigusi.
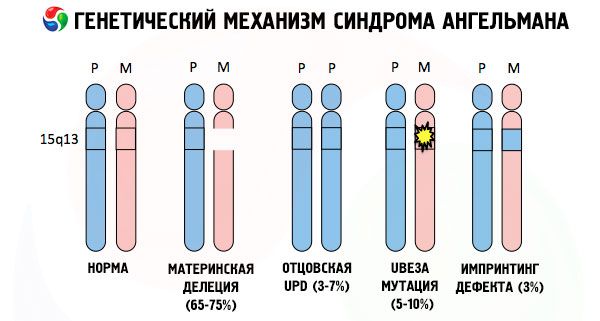
Selle haiguse puudub spetsiifiliste geenide emade kromosoomi kao või / vähenemisest aktiivsust üksikute geenide (enamikel juhtudel ube3a geeni metabolismis osalev Ubikitiin - valgulagundamises teiste regulatoorsete valkude). Selle tulemusena diagnoositakse lapsele vaimse arengu kõrvalekaldeid ja füüsilisi deformatsioone.
Sümptomid angelmaani sündroom
Angelmani sündroomi sümptomaatika mõjutab lapse elu ja arengu erinevaid aspekte: füüsilist, neuroloogilist ja psühholoogilist. Selle alusel saame eristada kolme sümptomite rühma, mis viitavad selle patoloogia arengule.
- Välised või füüsilised sümptomid:
- võrreldes tavalise suurusega pagasiruumi ja jäsemega ebaproportsionaalselt väike pea
- liiga suu,
- näol on peaaegu alati naeratus (avatud suuga);
- haruldased hambad
- kitsas ülahuus
- tihti kleepides läbi lai keele,
- väljaulatuv alumine lõualuu
- terav lõug,
- väga kerge nahk, sageli ka juuksed (albinism, mis on seotud asjaoluga, et keha ei toodeta pigmendi melaniini);
- tumedad täpid kerge naha (hüpopigmentatsioon ebapiisava melaniini tootmise tõttu)
- füüsilised või välised sümptomid: silmahaigused, nagu nägemisnärvi strabismus või atroofia,
- selgroosa kõverus (skolioos)
- jäigad jalad (kui jalgsi mees ei painuta oma põlvili tõttu liigeste vähest mobiilsust, siis võrdub see nukuteadiga).
- Vaimse ja emotsionaalse arengu sümptomid:
- vaimse arengu tugev lag,
- tarbetult emotsionaalne, mürarikas, uhke käitumine
- sagedane hooramine
- väljendatud sõbralikkus, mida rõhutavad pidevalt naeratus näole,
- sagedane, juhuslik naer.
- Neuroloogilised sümptomid:
- jäsemete treemor,
- liikumise ebapiisav kooskõlastamine tasakaalustamatusega,
- lihaste toonuse vähenemine
- mitmesugused unehäired,
- sagedased hüsteerilised krambid lapsepõlves,
- kõnehäired (laps hakkab hiljaks rääkima, tal on nõrk suhtlemisoskus ja nõrk kõne);
- hüperaktiivsus suurenenud erutusvõime taustal,
- kontsentratsiooni ja koolituse raskused.
Kuid see on haiguse üldine pilt. Tegelikult sõltub Anghelmani sündroomi kliiniline pilt suuresti haiguse arenguastmest ja patoloogia põhjustanud kromosomaalse mutatsiooni tüübist. See tähendab, et erinevatel patsientidel võib haiguse sümptomoloogia oluliselt erineda, mis pikka aega ei võimaldanud sarnase kliinilise pildi korral patoloogiat isoleerida.
Sümptomite koguarvu võib kindlaks teha selliselt, et see on kõigile eranditult patsientidele iseloomulik:
- vaimse arengu tõsised kõrvalekalded,
- ebapiisav käitumine (tarbetu naer, suurenenud erutavus, tähelepanelik kontsentratsioon, eufooria seisund);
- motoorsete oskuste vähearenenud
- kehaliste liikumiste koordineerimine, kõndimise atakia (ebaühtlane tempo, pööramine küljelt küljele jne), jäsemete treemor.
- kõneside rikkumine mitteverbaalsete kommunikatsioonivahendite ülekaaluga.
Suurtes enamuses patsientidel esinevate sümptomite hulgas võib eristada järgmisi nähte:
- ebaproportsionaalne pea ja pagasiruumid, mis on tingitud füüsilise arengu hilinemisest,
- paljudel patsientidel on kolju kuju selline, et aju suurus on väiksem kui tervetel inimestel (mikrotsefaalia),
- eakatel kuni kolmeaastased epileptilised krambid, kellel on eakatel tugevus ja sagedus järk-järgult vähenenud,
- EEG indeksite moonutused (võnked ja madalsageduslike lainete kõrge amplituud).
Need sümptomid ilmnevad üsna tihti, ent 20% -l Angelmanni sündroomiga patsientidest puuduvad nad.
Isegi harvemini saab diagnoosida haiguse selliseid ilminguid nagu:
- selgelt väljendatud või kergelt strabismus
- keele liikumise nõrk kontroll, mille tagajärjel patsiendid jätavad oma keele ilma põhjuseta
- neelamis- ja imemisraskused, eriti noorematel lastel,
- naha ja silmade pigmentatsiooni rikkumine,
- tõstetud või painutatud käte käes,
- giperperfleksia
- unehäired, eriti lapseeas
- sageli süljeeritus
- maha surutud janu
- liigselt aktiivsed närimiskummid
- ülitundlikkus kuumuse suhtes
- lameda peaga
- arenenud alumine lõualuu
- siledad peopesad.
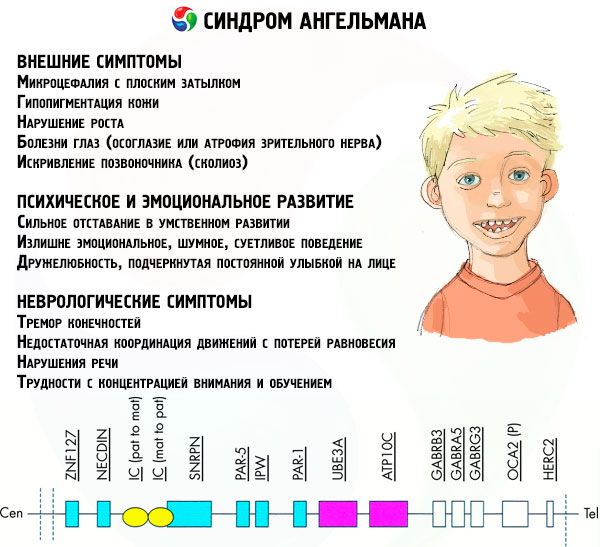
Üsna suurel protsendil patsientidest on urineerimisel probleeme, mida nad halvasti kontrollivad, trahvi motoorsete oskuste rikkumine, mis tekitab raskusi iseteeninduses ja koolituses, ülekaalulisus. Praktiliselt kõigil patsientidel algab puberteet hiljem kui terved eakaaslased.
Angelmani sündroomiga lapsed on suulise kõne mõistmisel ja mõistmisel head, kuid ei soovi vestluses osaleda, piirates nende kõnet kümneid igapäevaelus vajalikke sõnu. Kuid täiskasvanueas näivad sellised patsiendid nooremaid kui nende kaaslasi ilma geneetiliste patoloogiateta.
Paljud Angelmanni sündroomi sümptomid on jäigad, seega muutub haiguse kliiniline pilt vanusega. Krambid ja epilepsiahoogid muutuvad harvememaks või kaovad üldse, patsient muutub vähem ärritatuks, une lõpeb.
Tüsistused ja tagajärjed
Angelmani sündroom on tänaseni krooniline krooniline patoloogia, mis jätab patsientidelt võimaluse elada normaalset elu. Milline on beebi elu SA, sõltub suurel määral kromosoomide kõrvalekalde tüübist.
Kromosoomipiirkonna paljundamine enamikul juhtudel ei sobi kokku eluga. Ja isegi kui sellised patsiendid ei sure ei ole lapsekingades ega jõua puberteedi, ei ole neil laste olemasolu.
Anghelmani sündroomi geenide osa kustutamine või puudumine on enamasti takistuseks lapsele, kes õpivad kõndima ja rääkima. Sellistes laste puhul esineb vaimset alaarengut raskemas vormis, epilepsia-rünnakud sageli esinevad ja nende intensiivsus on palju tugevam kui teiste kromosomaalsete kõrvalekallete korral.
Kui on mutatsioon ühe geeni, võttes nõuetekohaselt arvesse ja lähenemisviisi, laps saab õpetada põhitõdesid iseteeninduse ja sidepidamine meeskonnas, kuigi see veel maha arengus eakaaslastega.
Angelmanni sündroomiga laste puhul on oma olemuselt heatahtlik põhiline vanemate armastus ja tähelepanu. Ainult sel juhul kannab lapse koolitus vilja, isegi väikseid. Muidugi ei saa tavakoolis olevad patsiendid SA-ga õppida. Neil on vajadus eriklasside järele, kus lastele õpetatakse kõigepealt keskenduma oma tähelepanu ning seejärel annavad nad järk-järgult kooliõppe põhitõdesid.
Diagnostika angelmaani sündroom
Angelmani sündroom on kaasasündinud patoloogiline areng. Kuid mõne olukorra tõttu ei ole diagnoosimine seda lapsepõlves ja varases lapsepõlves kõige sagedamini võimalik. Selle põhjuseks on mittespetsiifilisus ja kergeid sümptomeid imikutele ja väikelastele kuni 3 aasta jooksul. Ja meie riigis haiguse esinemissagedus ei ole nii suur, et arstid on õppinud tundma seda oma sarnasuse poolest.
Angelmani sündroom imikutel võib avalduda kujul vähendatud lihastoonust, mis avaldub kujul toitmisprobleemid (nõrkus imemiseks ja neelamisfunktsioon) ja hiljem õpiraskused kõndides (nt laste palju hiljem alustada jalgsi). Need sümptomid on esimesed kõrvalekalded märke lapse arengust, mis võib olla seotud kromosoomide kõrvalekaldega. Kinnitage, et see eeldus võib olla ainult geneetiline analüüs.
Erilist tähelepanu pööratakse lastele, kelle vanematel on erinevad genoomilised või kromosoomide kõrvalekalded. Lõppude lõpuks ei saa haigus mingil viisil avalduda, ja kui patoloogia ilmneb õigeaegselt, alustades lapsega kõvasti tööd, on koolituses võimalik saavutada palju suuremat edu, haiguse progresseerumise aeglustamisel.
Kui vanematel on erinevad kromosomaalsed kõrvalekalded, tehakse geneetiline analüüs isegi enne lapse sündi, sest CA on üks patoloogiatest, mida saab embrüonaalses seisundis tuvastada.
Geneetiliste uuringute materjalide kogumist saab teha kahel viisil:
- invasiivne (teatud riskiprotsent, kuna see peab sisenema emakasse amnionivedeliku testimiseks);
- mitteinvasiivne (lapse DNA analüüs emalt verest).
Siis viiakse läbi järgmised uuringud:
- in situ hübridisatsiooni fluorestsents (FISH-meetod) - DNA-sond, mis on märgistatud spetsiifilise värvainega uuritavasse DNA-sse, millele järgneb mikroskoopiline uurimine.
- mutatsioonide analüüs ube3a geenis ja geenide trükkimine,
- DNA metüülimise analüüs geneetikas kasutatavate erimeetodite abil.
Geneetilised analüüsid annavad kromosomaalsete kõrvalekallete kohta üsna täpset teavet, nii et tulevased vanemad teavad ette, millised nad peaksid olema valmis. Sellest hoolimata on erandeid. Teatud patsientide grupis on kõigi sümptomite näitajatest tingitud sümptomite ilmnemisel analüüside tulemused normaalsed. Ie. Patoloogia avastamiseks on võimalik lapsega varakult jälgida vaid hoolikalt: kuidas sööb, kui ta hakkas kõndima ja rääkima, kas jalad kõnnivad kõndides jne.
Lisaks FISH-meetodil hulgas meetodid diagnoosivahend Angelmani sündroom võib eristada tomograafia (CT või MRI), aidates olekut kindlaks ja aju suuruse ja elektroentsefalogramme (EEG), mis näitab, kuidas üksikud osad aju töötab.
Arstide lõplik diagnoos määratakse tavaliselt 3-7 aasta vanuseks, kui patsiendil on juba enamus sümptomeid ja haiguse arengu dünaamika on nähtav.
Millised testid on vajalikud?
Diferentseeritud diagnoos
Angelmani sündroom on geneetiline patoloogia, millel ei ole konkreetset ilmingut. Enamik sümptomeid võib võrdselt näidata nii CA-d kui ka muid geneetilisi haigusi.
Anghelmani sündroomi diferentseeritud diagnoosimisel kasutatakse järgmisi patoloogiaid:
- Pitt-Hopkinsi sündroom (patsientidel on vaimne alaareng, rõõmsameelne iseloom, naeratavad, neil on suhteliselt suur ja lai suu, märgitakse mikrotsefaalia). Erinevus - hüperventilatsiooni rünnakud ja äratõukereaktsiooni hingamine.
- Kristiansona sündroom (patsiendid, kes on vaimselt aeglustunud, kellel on rõõmsameelne käitumine, võimetus rääkida, neid iseloomustab mikrotsefiaal, ataksia, krambid, lihaste tahtmatud liikumised).
- Mowata-Wilsoni sündroom (sümptomid: vaimne aeglustumine, epilepsia krambid, terav lõug, avatud suu, näo õnne avaldumine, mikrotsefaalia). Erinevus on suur vahemaa silmade vahel, silmad asetsevad sissepoole, nina ots on ümar, aurikleik pööratakse tagasi.
- Kabuki sündroom (iseloomustab kerge kuni mõõduka vaimse alaarenguga, kõnehäired ja motoorseid oskusi, lihasnõrkus, krambid, mikrotsefaalia suured lõhed zudami, koordinatsioonihäired). Erinevus - kaela kujuline kulm, alumiste silmalaugude ümberpööratud külgosa, laiad silmad, pikad silmade lõiked pikkade jämedate ripsmetega.
- Rett-sündroom (eristamine CA-ga naistel). Sümptomid: hilinenud kõne areng, konvulsioonilised krambid, mikrokefalüüs. Erinevus - näol ei ole õnnelikku väljendust, on apnoe ja apraksia rünnakud, mis lõpuks edenevad.
- Sündroom autosoomne retsessiivne vaimse tardatsii 38 (sümptomid: vaimne alaareng koos märgatavat viivitust arengut motoorseid oskusi ja kõne, lihasnõrkus, toitmisprobleemid imikueas, impulsiivsus). Erinevus on iirise sinine värv.
- Geeni MESR 2 mitmekordse sündroomi sündimine (diferentseerumine SA-ga meestel). Sümptomid: raske vaimne alaareng, lapsepõlvest tingitud lihaste nõrkus, kõne või selle puudumine, epilepsia. Erinevused - progresseeruv müopaatia, pidevalt korduvad infektsioonid.
- Klifstra sündroom (sümptomid: kõnehäired ja mõtlemine, lihasnõrkus, unehäired, vähene tähelepanu, avatud suuga, hüperaktiivsus, krambid, ataksia, tasakaalu häired). Erinevused - lamedad näod, lühike nurk nina, laiad silmad, suur ümberpööratud alumine äär, agressiivsed rünnakud.
- Smith-Magenis'i sündroom (mida iseloomustavad krambid, unehäired, intellektuaal- ja motoorse arengu häired). Erinevused - lai ja lame nägu, kumer otsmik.
- Kulena-de Vriesi sündroom (kerge ja mõõdukas vaimne aeglustumine, lihasnõrkus, konvulsioonirünnakud, sõbralikkus). Erinevused - pikk nägu, millel on suur otsmik, väljaulatuvad kõrvad, kaldkilbid, liigeste liikuvus, kaasasündinud südame patoloogia.
- Syndrome Philan - McDermid (sümptomid: vaimne alaareng, kõnepuudulikkus või selle puudumine). Erinevused - suured käed arenenud lihastega, lihaste nõrkus alates sünnist, nõrk higistamine.
Angelmani sündroomi sarnaseid sümptomeid saab "hooplemises" ja kuidas sellist patoloogiat adenilsuktsinazy defitsiit, autosoomne retsessiivne sündroom vaimse alaarengu 1, kromosoomi 2q23.1 dubleerimist sündroom, haplopuudulikkusega geenide FOXG1, STXBP1 või MEF2C ja teised.
Arsti ülesanne on teha täpset diagnoosi, diferentseerida Angelmanni sündroomi sarnaste sümptomitega patoloogiatega ja määrata efektiivne ravi, mis on oluline diagnoositud haiguse arengu jaoks.
Kellega ühendust võtta?
Ravi angelmaani sündroom
Angelmani sündroom viitab nende patoloogiate kategooriale, mille otsimine ravimi efektiivseks raviks on selle päevaga seotud. Haiguse etioloogiline ravi on erinevate meetodite ja vahendite väljatöötamisetapis, millest paljud ei ole veel inimestel testitud. Seni arstid olema piirata sümptomaatilise ravi, et aidata kuidagi leevendada nende laste ja täiskasvanute nuku sündroom, kannatavad epilepsiahoogude, süljeeritus, hüpotensioon ja unehäirete.
Seega vähendage epilepsiavastaste krampide sagedust ja tugevust koos õigesti valitud krambivastaste ravimitega. Probleem on aga selles, et krampide raviks CA erinevad tavalistest epilepsiahoogude et neid iseloomustab mitut liiki krampide ja seega seisundi leevendamiseks on kasutuselevõtuga paljude ravimitega.
Kõige populaarsem krambivastast raviks kasutatav Angelmani sündroom on: valproehappe, topiramaadi, lamotrigiini, levetiratsetaami clonazepam ja põhinevad preparaadid. Harvem kasutatakse ravimeid põhinevad karmazepina, fenütoiin, fenobarbitaal, etosuksimiid, sest mõned neist võivad käivitada paradoksaalse efekti, on tugevdada ja sageduse suurendamiseks epileptilised krambid. See juhtub, kui ravimit kasutatakse monoteraapiana.
Süljeerituse raviks kasutatakse tavaliselt kahte meetodit: ravimit (sülje moodustumist pärssivad preparaadid) ja operatiivset, mis seisneb süljesortide reimplantatsioonis. Kuid CA puhul on need meetodid ebaefektiivsed ja küsimus jääb avatuks. Vanemad ja need, kes selliseid patsiente hooldavad, peame selle hetkeni erilist tähelepanu pöörama, kuna patsiendid ise tavaliselt ei kontrolli süljeerumist ja mõned lihtsalt ei saa ennast hoolitseda.
Teine probleem on une lühike kestus. Sageli ei ela Angelmani sündroomiga lapsed enam kui 5 tundi, mis mõjutab negatiivselt kogu organismi tööd. Hämmastajad, aktiivsed lapsed, armastavad mängud ja suhtlemine (isegi kui nad püüavad piirduda mitteverbaalsete viisidega) on päevast märkimisväärselt väsinud. Hea puhkuse saamiseks vajab keha täiuslikku unistust, kuid see on lihtsalt selle probleem.
Tundub, et parandada une ärritatavates patsientidel peaks olema piisavalt ravimeid sedatiivset toimet (fenotiasiinid ja atüüpilised antipsühhootikumid), rahustab närvisüsteemi. Kuid CA puhul on selliste ravimite kasutamine täis negatiivsete mõjude ilmnemist. Seetõttu arstid eelistavad endiselt väikesed uinutite, nagu "Melatoniin" (loomuliku hormonaalse preparaadi põhjal une hormooni), mis annavad patsientidele tund enne magamaminekut 1 tablett, ja "Diphenhydramine". Arst määrab manustamise sageduse ja annuse, sõltuvalt patsiendi seisundist ja vanusest.
Mõnikord on ingelmani sündroomiga patsientidel seedehäired ja väljaheide. Tooli reguleerimine on võimalik lahtistavate preparaatide abil (see on parem kui fütogenees).
Ja sa võid läheneda probleemi erinevalt, nagu seda tegi Ameerika arstid, põhineb mõned ravimeetodid autism, sest paljud sümptomid iseloomulik SA, on omane ka autism (impulsiivsus, tahtmatud liigutused, korduv meetmeid, tähelepanu puudulikkuse, probleeme side jne .) On täheldatud, et manustamine hormoon sekretiin, normaliseerida seedimist ja tool, positiivne mõju patsientide tähelepanu ja oksütotsiin aitab parandada laste kognitiivsed võimed ja mälu, parandada käitumist.
Tõsi, mõned hormoonid on siin hädavajalikud, eriti laste puhul. Angelmani sündroom näitab käitumuslikku ravi, tööd psühholoogi ja logopeediga (õpetades mitteverbaalseid suhtlusviise ja viipekeelt). Selliste laste koolitamine peaks põhinema individuaalsel programmil, kus osalesid spetsiaalselt koolitatud õpetajad, psühholoog ja vanemad. Kahjuks pole see kõikjal võimalik ning pered jäävad oma probleemiga üksi.
Kuna paljudel väikestel CA-ga põdevatel patsientidel on madal lihastoonus ja liigeseprobleemid, pööratakse suurt tähelepanu füsioterapeutiline ravi. Enamasti kasutavad arstid parafiinirakenduste, elektroferoosi, magnetoteraapiat.
Aktiivne toonimassaaž ja spetsiaalsed füsioteraapia harjutused aitavad haigeil lapsel mõnda aega kindlalt jalgadele ja kõndides seista. Sellega seoses on eriti kasulik vesiviljelus, mida CA soovitab kasutada jahedas vees. See suurendab lihaste toonust ja õpetab lapsel oma keha omama, koordineerima liikumisi.
Krambivastane ravi
Anghelmani sündroomi kõige ohtlikum sümptom on krambid, mis sarnanevad epilepsiahoogudega. Seda sümptomit täheldatakse 80% patsientidest, mis tähendab, et neile kõigile tuleb anda efektiivne krambivastane ravi.
Epilepsiahoogude ravi viiakse läbi vitamiinide ja antikonvulsantide abiga. Kui Angelmani sündroom, millega kaasneb Convulsive sündroom, on kasulik vitamiine B rühma, samuti vitamiine C, D ja E. Aga vitamiiniravi määrata oma antud juhul on väga ohtlik, sest kontrollimatu vitamiinide võib vähendada tõhusust epilepsiavastaste ravimite ja provotseerida uue, raske ja pikaajaline krambid.
Krambivastaste ravimite valikut ja nende efektiivse annuse määramist peaks käitlema ka spetsiaalne arst. Samuti otsustab ta, kas piisab ühest ravimist või kui patsient peab võtma pikka aega kaks või enam ravimit.
Enamik patsiente arstid kirjutada narkootikumide valproehappe ( "valproehappe", "Depakinum", "Konvuleks", "valparin" jt.), Mis takistavad krambid, parandada meeleolu ja vaimse seisundi patsientidel.
Valproehape on saadaval tablettide, siirupi ja süstitavate lahuste kujul. Kõige populaarsem ravim on pikaajalise toimega ravim "Depakin" tablettidena ja veenisiseseks manustamiseks mõeldud lahusena. Ravimi annust määrab arst eraldi, olenevalt patsiendi kehakaalust, vanusest ja seisundist.
Võtke ravimit toiduga 2-3 korda päevas. Keskmine päevaannus on 20-30 mg ühe kilogrammi patsiendi kehakaalu kohta, maksimaalne annus on 50 mg / kg päevas.
Vastunäidustused. Seda ei kasutata maksa- ja kõhunäärmehaiguste, hemorraagilise diatsesi, hepatiidi, porfüüria ja ülitundlikkuse ravimiseks.
Kõrvaltoimete hulka võib kuuluda käte värisemine, seedimine ja väljaheide, kehakaalu muutused.
"Topiramaat" on ka CA-s valitud ravim. See on valmistatud tablettide kujul ja seda kasutatakse monoteraapiana ja kombinatsioonis teiste ravimitega.
Kasutusmeetod ja annus. Võta pillid sees, sõltumata toidu tarbimisest. Täiskasvanute esialgne ööpäevane annus on 25-50 mg, lastele 0,5-1 mg / kg. Iga nädala järel suurendatakse annust vastavalt arsti ettekirjutusele.
Ravimit ei tohi kasutada raseduse ja imetamise ajal, samuti selle koostisosade tundlikkuse suurenemisel. Ravimil on palju erinevaid kõrvaltoimeid.
Drugs, mida arst saab ette temperatuuril Angelmani sündroom "Klomazepam", "Rivotril'iga" "Lamotrigiinil", "Seyzar", "Lamictal", "levetiratsetaami", "Keppra", "Epiterra" jt.
Alternatiivne ravi ja homöopaatia
Alternatiivne meditsiin, nagu homöopaatilised ravimid, kindlasti erineb võrdlemisest ohutusest, kuid siin on Angelholmi sündroomiga seoses sellise ravi efektiivsust pidada vastuoluliseks.
Kuigi mõni alternatiivne ravi võib ikkagi aidata. See on epilepsiahoogude peatamine. Sellega seoses võib taimteraapia olla üsna tõhus.
Hea efekt on tingitud pojengist, lagrillist ja hiivlast põhinevast ravimitasust (komponente võetakse võrdsetes kogustes). Rööpad tuleb jahvatada jahuks. Pärast 2 nädala möödumist vastuvõtuse algusest saad märkida konvulsioonirünnakute sageduse märkimisväärse vähenemise.
Kasulik krampide ja leivendite keetmise jaoks (1 tl klaasi keeva veega). Preparaati keedetakse 5 minutit ja nõutakse poole tunni jooksul. Võtke ravim üleöö 14 päeva jooksul.
Epilepsiavastaste rünnakute jaoks efektiivseks peetakse vett (või alkoholi) infusioonimürki.
Alates homöopaatia krampide vastaseid ravimeid koos Angelmani sündroom võib kasutada ravimeid, mis põhineb kummel ja motherwort, Acidum hydrocyanicum, Argentum nitricum, kaalium bromatum, Arsenicum album. Kuid tuleb arvestada, et efektiivseid ja ohutuid preparaatide annuseid võib igal konkreetsel juhul nimetada ainult arstiga, kes on homöopaat.
Ärahoidmine
Nagu lugejat on ilmselt juba mõistnud, et vältida geenide ja muude kromosoomide kõrvalekallete mutatsiooni, on meditsiin ikkagi jõu kaugel ja ka olukorra parandamiseks. See võib kõigile juhtuda, kuna Angelmanni sündroomiga lapsed sünnivad ka tervetel vanematel ja geneetika, mis hetkel on üks vähemarenenud teadusharusid, ei suuda seda veel selgitada.
Ainus asi, mida on võimalik teha, on võtta vastutus raseduse planeerimisele, registreerida ja õigeaegselt läbi vaadata. Kuid jällegi ei peaks selline meede pigem profülaktilisi kui kognitiivseid, nagu iga uuringut. Kuid noored vanemad teavad ette, milliseid ettevalmistusi teha ja kui nad annavad positiivse vastuse, otsustavad nad, kas neil on võimalik võtta selline vastutus haige lapse kasvatamise eest.
Prognoos
Anghelmani sündroomi prognoos sõltub kromosoomide kõrvalekaldumise olemusest ja selle avastamise õigeaegsusest. Kõige raskem osa on nende laste jaoks, kelle 15 kromosoomil on "puuduvad" geenid (kustutatud). Sellistes patsientides kõndimise ja rääkimise tõenäosus on äärmiselt väike. Ülejäänud juhtudel, kus on tähelepanelik lähenemine ja armastus teie lapsega, on võimalik korrigeerida.
Sellised patsiendid, paraku, ei saa ühiskonna täisliikmeks, hoolimata asjaolust, et nad pole kaugelt rumalad, mõistavad nad kõnet ja selle tähendust. Siin on lihtsalt kommunikatsiooniprobleemid, mis neil on kogu elu jaoks. Patsiente saab lapsepõlves kirjutada viipekeele, kuid neid ei saa sundida suhelda sõnadega. Rääkivate patsientide sõnavara piirdub vähemalt igapäevases kasutuses olevate sõnadega (5-15 sõna).
Mis puudutab Anghelmani sündroomiga patsientide oodatavat eluiga ja üldist tervislikku seisundit, siis need joonised kõikuvad keskmiselt. Täiskasvanueas seisavad patsiendid üldiselt silmitsi terviseprobleemidega, nagu näiteks skolioos ja ülekaalulisus, mis õige lähenemisega ravile ei ole eluohtlikud.