
Tricher Collinsi sündroom
Viimati vaadatud: 23.04.2024

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.

Kui loote luude arengut häired protsessid olla tõsised kraniofatsiaalse deformatsioone ja üks sortide sellise patoloogia Treacher Collins sündroom (TCS) või mandibulofastsialny, st maxillofacial düsostoos.
Haiguskood vastavalt ICD 10: XVII klass (kaasasündinud väärarengud, deformatsioonid ja kromosoomide kõrvalekalded), Q75.4 - mandibulofatsiaalne düsoosoos.
Põhjused tricher Collinsi sündroomist
See sündroom oli nime saanud väljapaistva Briti silmaarst Edward Tricher Collinsi poolt, kes kirjeldas patoloogia põhijooni enam kui sada aastat tagasi. Kuid Euroopa arstid sageli nimetame seda liiki kõrvalekaldeid näoluude ja lõualuu või sündroom FRANCESCHETTI - põhineb põhjalikke uuringuid Šveitsi silmaarsti Adolf FRANCESCHETTI, kes vermis termini "mandibulofastsialny düsostoos" keset eelmisel sajandil. Meditsiiniline kogukond kasutab ka nimetust - Franceschetti-Collinsi sündroom.
Põhjustab Treacher Collins sündroom - geenimutatsioonid TCOF1 (locus 5q31.3-33.3 kromosoomid), mis kodeerib nucleolar fosfoproteiin, mis vastutab moodustamise kraniofatsiaalse osa inimese embrüo. Selle valgu koguse enneaegse languse tagajärjel häirib biogeenne ja rRNA funktsioone. Vastavalt geneetikud Human Genome Research Program, need protsessid tagajärjel väheneb vohamises embrüonaalsete neuraalharjast rakud - rullil piki närvi küveti, mille käigus arengus embrüo neuraaltoru sulgumist.
Kihistu esiosa kolju koe põhjustatud muundumine ja rakkude diferentseerumise pealise (pea) osa neuraalharjast et rändavad mööda neuraaltoru esimesele ja teisele branchial kaared embrüo. Nende rakkude defitsiit põhjustab ka kõhuõõne deformatsioone. Anomaaliate esinemise kriitiline periood on 18 kuni 28 päeva pärast viljastamist. Lõpetamisel rände neuraalharjast rakud (neljandas rasedusnädalani) on moodustatud peaaegu kõik lahti Mesenhümaalsed koe nägu, mis hiljem (5 kuni 8 nädalat) diferentseeruvad skeleti ja sidekoe kõigi osade nägu, kael, kurk, kõrva (sealhulgas sisemine) ja tulevased hambad.
Pathogenesis
Patogenees Treacher Collins sündroomi enamasti perekondlikud ja kõrvalekalle päritav autosoomne dominantne põhimõtet, kuigi on juhtumeid autosoomne retsessiivne edastamise vead (mutatsioonid teistes geenides, eelkõige POLR1C ja POLR1D). Kõige ettearvamatu maxillofacial düsostoos on see, et mutatsioon on päritud lastele ainult 40-48% juhtudest. See tähendab, et 52-60% patsientidest põhjustab Treacher Collins sündroom ei seostata juuresolekul kõrvalekaldeid perekonna ning neil arvatakse olevat patoloogia tekib juhuslik geneetilisi mutatsioone de novo. Uued mutatsioonid näitavad tõenäoliselt teratogeenseid toimeid lootele raseduse ajal.
Põhjused Selle sündroomi teratogeenne eksperdid nimetavad suurtes annustes etanoolis (etüülalkohol), kiiritus, sigaretisuits tsitomegavirus ja toksoplasmoosi, samuti herbitsiidid glüfosaadi (Raundal, Glifor, Tornado ja teised.). Ja nimekiri iatrogeensele tegurid olid preparaatide seborröa ja akne 13-cis-retinoehape (isotretinoiini akutaan); krambivastane ravim fenütoiin (dilantiin, epanutiin); psühhotroopsed ravimid diasepaam, valium, relanium, Seduksen.
Sümptomid tricher Collinsi sündroomist
Enamasti sõltuvad mandibulofastilise düsoostoosi kliinilised tunnused ja nende raskusaste geenimutatsioonide manifestatsioonide omadustest. Ja esimesed selle anomaaliumi tunnused on enamikul juhtudest nähtavad lapsest kohe pärast tema sündi: nägu Tricher Collinsi sündroomiga on iseloomulik. Lisaks on morfoloogilised kõrvalekalded tavaliselt kahepoolsed ja sümmeetrilised.
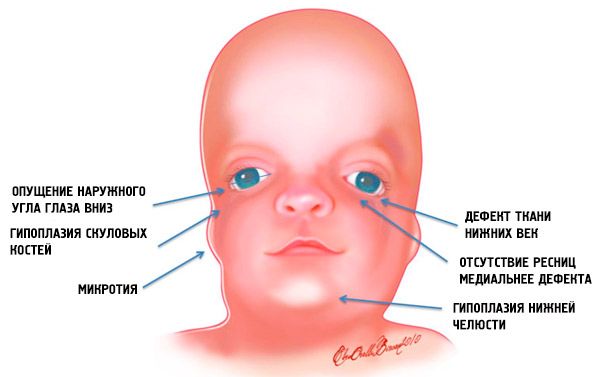
Kõige ilmsemad Tricer Collinsi sündroomi sümptomid on:
- alaarengut (hüpoplaasia) näoluude kolju: väljaulatuvate, väljaulatuvate protsessi otsmikuluus külgmised Pterygoid plaatidel, ninakõrvalkoobaste, alalõua luude epifüüsiga ja väljaulatuvad (kondüülid);
- alumiste lõualuude luude vähene areng (mikrognaatiia) ja tihedam kui tavaliselt mandibulaarne nurk;
- nina on normaalse suurusega, aga see tundub suurepärane, kuna hüpoplasia on ülekaalulised kaared ja nõrkade arkade puudumine või halvenemine templite piirkonnas;
- silmade pilud on kahanevad, see tähendab, et silmade sisselõige on ebanormaalne, kusjuures välimine nurgad on langetatud allapoole;
- alumiste silmalaugude (koloboma) defektid ja nende ripsmete osaline puudumine;
- ebaregulaarse kuju kõrvad laia hälvetega, kuni nende paiknemiseni alumises lõualuumis, pooride puudumine, pimedad fistulid kitse kõrva ja suu nurga vahel jne;
- kitsenev või infektsioon (atresia) ja kõrvakeskmete luulude kõrvalekalded;
- paisteli süljenäärmete puudumine või hüpoplaasia;
- neelu hüpoplaasia (neelu ja hingamisteede kitsenemine);
- kõva mao mittehävitamine (hunt suhu), samuti pehme salli puudumine, lühenemine või liikumatus.
Sellistel anatoomilistel kõrvalekalletetel on kõigil juhtudel tüsistused. Need on funktsionaalsed kuulmiskahjustused juhtivate (juhtivate) kuulmiskadude või täieliku kurtusena; nägemise halvenemine silmapilguse ebaõige moodustumise tõttu; Suulise vea defektid põhjustavad raskusi söötmise ja neelamisega. Hammaste ummistuse (äärmiste hammustustega) tekkega on seotud lõualuu, mis omakorda tekitab probleeme närimise ja liigesedega. Pehme palmi patoloogiad selgitavad nina häält.
Tüsistused ja tagajärjed
Tagajärjed näokolju anomaaliad Treacher Collins sündroom, mis avaldub selles, et pärast sündi oma tavapärase intellektuaalse võime, kuid kuna kuulmine vead ja muud haigusseisundid teisese vaimne alaareng.
Lisaks on selliste defektidega lapsed teadlikud nende alaväärtusest ja kannatustest, mis mõjutab negatiivselt nende närvisüsteemi ja psüühikat.
Diagnostika tricher Collinsi sündroomist
Tricher Collinsi sündroomi postnataalne diagnoos põhineb peamiselt kliinilistele nähtudele. Maksaiguse ja kauakeste düsoostoosi saab kergesti kindlaks määrata koos sündroomi täieliku väljendusvõimega, kuid kui patoloogia sümptomid on minimaalsed, võivad õige diagnoosi sõnastamisega tekkida probleeme.
Sellisel juhul tuleb erilist tähelepanu pöörata kõigi kõrvalekalletega seotud funktsioonide, eriti hingamisteid mõjutavate funktsioonide (uneapnoe ohu tõttu) hindamisele. Samuti viiakse läbi hemoglobiini söötmise ja hapniku küllastatuse efektiivsuse hindamine ja jälgimine.
Tulevikus - 5.-6. Päeval pärast sünnitust - on vajalik teada saada kuulmise kahjustuse määr audioloogiliste testide abil, mis tuleks läbi viia sünnitushaiglas.
Tehakse eksam, mille käigus instrumentaalne diagnoos viiakse läbi kolju-näo düsmorfoloogia fluoroskoopia abil; pantomograafia (näo kolju luustiku struktuuride panoraamröntgen); täieliku kolbiarvuga kompuutertomograafia erinevates projektsioonides; CT või MRI aju, et määrata kindlaks sisemise kuulmismeetodi seisund.
Varaseim - Sünnieelne - diagnoos näokolju anomaaliad juuresolekul Treacher Collins sündroom, perekonna ajalugu on võimalik koorioni hattude valimi 10-11 rasedusnädalal (protseduuri ähvardab nurisünnitus ja nakkuse emakas).
Samuti võetakse pereliikmete vereanalüüsid; 16-17 rasedusnädalal võetakse amniootilise vedeliku (transabdominaalne amniotsenteede) analüüs; raseduse 18-20 nädala tagant tehakse fetoskoopia ja võetakse verd platsentaarbarakku.
Kuid kõige sagedamini selle sündroomi prenataalses diagnoosis kasutab loote ultraheli (raseduse 20-24 nädala jooksul).
Millised testid on vajalikud?
Diferentseeritud diagnoos
Need samade meetoditega eksperdid kasutavad vajadusel diferentsiaaldiagnoosimist, tunnustada tasakesi väljendunud Treacher Collins sündroom ja eristada seda teistest kaasasündinud väärarengute kraniofatsiaalse luud, eelkõige: sündroomide Apert, Crouzon, Nagera Peters-Hevelsa, Hellerman-Staefa, samuti koos hemifacial microsomia (Goldenhar sündroom), hüpertelorism, varase imperforate kolju õmblusniitide (craniostenosis) või rikkudes fusiooni näoluude (craniosynostosis).
Kellega ühendust võtta?
Ravi tricher Collinsi sündroomist
Nagu kõikidel geneetiliselt konditsioneeritud sünnidefektetel, on Tricer Collins'i sündroom raskete vormide korral vaid palliatiivne, kuna selliste patoloogiate puhul pole lihtsalt ravimeetodeid. Selle sündroomi spektri ja deformatsioonide ulatus on ulatuslik ja järelikult on meditsiinilise sekkumise laadil ja intensiivsusel ka mitmeid valikuid.
Kuuldeaparaate kasutatakse kuulmise parandamiseks ja parandamiseks, kõneprofiili parandamiseks koos kõnespetsialistiga.
On vajalik kirurgiline sekkumine varases eas, rasketel juhtudel hingamisteede ahenemine (trahheostoomia.Ventilaatori läbi) ja kõri (läbi gastrostommiya söötmise) võib samuti vajada kirurgilist korrigeerimist maitse.
Operatsioonid alumiste lõualuude pikendamiseks viiakse läbi 2-3 aastat vanuses või vanemas eas. Pehmete kudede rekonstrueerimine hõlmab alumiste silmalaugude kolooniate ja aurikele plastiku parandamist.
Prognoos
Mida saab selle patoloogia jaoks prognoosida? See sõltub deformeerumise astmest ja sümptomite intensiivsusest. Tricker Collinsi sündroom on eluaegne diagnoos.
 [25]
[25]