Artikli meditsiiniline ekspert

Uued väljaanded
Charcot-Marie-Tooth'i haigus.
Viimati vaadatud: 12.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
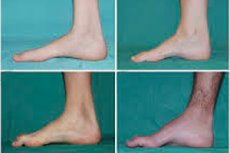
Peroneaalne lihasatroofia ehk Charcot-Marie-Toothi sündroom või haigus on krooniliste pärilike haiguste rühm, millega kaasneb perifeersete närvide kahjustus.
RHK-10 kohaselt on närvisüsteemi haiguste osas selle haiguse kood G60.0 (pärilik motoorne ja sensoorne neuropaatia). See kuulub ka harvaesinevate haiguste loetellu.
Epidemioloogia
Kliinilise statistika kohaselt on kõigi Charcot-Marie-Tooth'i tõve tüüpide levimus 100 000 elaniku kohta 19 juhtu (teiste allikate kohaselt üks juhtum 2,5–10 000 elaniku kohta).
CMT 1. tüüp moodustab umbes kaks kolmandikku juhtudest (üks juhtum 5000–7000 elaniku kohta) ja ligi 70% neist on seotud PMP22 geeni duplikatsiooniga. Seda tüüpi haiguse all kannatab üle maailma üle 1,2 miljoni inimese.
CMT 4. tüübi esinemissagedus on hinnanguliselt 1–5 juhtu 10 000 lapse kohta. [ 1 ]
Põhjused Charcot-Marie-Tooth'i haigus
Polüneuropaatiliste sündroomide klassifikatsiooni kohaselt viitab peroneaalne (fibulaarne) lihasatroofia, Charcot-Marie-Toothi neuraalne amüotroofia või Charcot-Marie-Toothi tõbi (lühendatult CMT) geneetiliselt määratud motoor-sensoorsetele polüneuropaatiatele. [ 2 ]
See tähendab, et selle esinemise põhjused on geneetilised mutatsioonid. Ja sõltuvalt geneetiliste kõrvalekallete olemusest eristatakse selle sündroomi peamisi tüüpe või liike: demüeliniseeriv ja aksonaalne. Esimesse rühma kuulub Charcot-Marie-Toothi tõbi tüüp 1 (CMT1), mis tekib PMP22 geeni duplikatsiooni tõttu 17. kromosoomis, mis kodeerib transmembraanset perifeerset müeliinivalku 22. Selle tulemusena tekib aksoniümbrise (närvirakkude jätkete) segmentaalne demüelinisatsioon ja närvisignaali juhtivuse kiiruse vähenemine. Lisaks võivad mutatsioonid esineda ka mõnes teises geenis.
Aksonaalne vorm on Charcot-Marie-Toothi tõve tüüp 2 (CMT2), mis mõjutab aksoneid endid ja on seotud patoloogiliste muutustega MFN2 geenis lookuses 1p36.22, mis kodeerib membraanivalku mitofusiin-2, mis on vajalik mitokondrite liitmiseks ja funktsionaalsete mitokondriaalsete võrgustike moodustumiseks perifeersete närvirakkude sees. CMT2-l on üle tosina alatüübi (mutatsioonidega spetsiifilistes geenides).
Tuleb märkida, et praegu on tuvastatud üle saja geeni, mille pärilik kahjustus põhjustab Charcot-Marie-Toothi tõve erinevaid alatüüpe. Näiteks RAB7 geeni mutatsioonid viivad CMT 2B tüübi tekkeni; SH3TC2 geeni (mis kodeerib ühte Schwanni rakkude membraanvalku) muutus põhjustab CMT 4C tüüpi, mis avaldub lapsepõlves ja mida iseloomustab motoorsete ja sensoorsete neuronite demüelinisatsioon (selle haiguse 4. tüüpi vorme on tosinat ja poolt).
Haruldane 3. tüüpi CMT (nimetatakse Dejerine-Sottase sündroomiks) hakkab arenema varases lapsepõlves ja selle põhjustavad mutatsioonid PMP22, MPZ, EGR2 ja teistes geenides.
Kui 5. tüüpi CMT tekib 5–12-aastaselt, ei täheldata mitte ainult motoorset neuropaatiat (alajäsemete spastilise parapareesi kujul), vaid ka nägemis- ja kuulmisnärvide kahjustusi.
Lihasnõrkus ja nägemisnärvi atroofia (koos nägemiskaotusega) ning tasakaaluprobleemid on CMT 6 tüüpi tüüpilised tunnused. Ja Charcot-Marie-Toothi tõve 7. tüübi korral ei esine mitte ainult motoor-sensoorset neuropaatiat, vaid ka võrkkesta haigus pigmentretiniiti kujul.
Meeste seas sagedasem on X-kromosoomiga seotud CMT ehk Charcot-Marie-Toothi haigus koos jäsemete tetrapareesiga (mõlema käe ja jalgade liikumisnõrkus). See on demüeliniseeriv tüüp ja arvatakse, et see tuleneb X-kromosoomi pikal harul asuva GJB1 geeni mutatsioonist, mis kodeerib konneksiin 32, Schwanni rakkude ja oligodendrotsüütide transmembraanset valku, mis reguleerib närvisignaalide ülekannet. [ 3 ]
Riskitegurid
KMT peamine riskitegur on haiguse esinemine perekonnas, st lähisugulastel.
Geneetikute sõnul on Charcot-Marie-Toothi haiguse autosomaalselt retsessiivse geeni kandjate puhul lapse haigestumise risk 25%. Ja lapse risk, et ta on selle geeni kandja (kuid tal pole mingeid sümptomeid), on hinnanguliselt 50%.
X-kromosoomiga seotud pärandumise korral (kui muteerunud geen asub naise X-kromosoomis) on 50% risk, et ema annab geeni edasi oma pojale, kellel tekib CMT. Haigus ei pruugi tekkida tüdruku sündides, kuid tütre pojad (lapselapsed) võivad defektse geeni pärida ja haigus areneb välja.
Pathogenesis
Mis tahes Charcot-Marie-Toothi haiguse korral on selle patogeneesi põhjuseks perifeersete närvide pärilik anomaalia: motoorne (liikumis-) ja sensoorne (tundlik).
Kui CMT tüüp on demüeliniseeriv, siis perifeersete närvide aksoneid kaitsva müeliinkesta hävimine või defekt viib närviimpulsside ülekande aeglustumiseni perifeerses närvisüsteemis - aju, lihaste ja meeleelundite vahel.
Haiguse aksonaalse tüübi korral mõjutavad aksonid ise, mis mõjutab negatiivselt närvisignaalide tugevust, mis ei ole piisav lihaste ja meeleelundite täielikuks stimuleerimiseks.
Loe ka:
Kuidas Charcot-Marie-Toothi sündroom edasi kandub? Defektsed geenid võivad pärida autosomaalselt dominantsel või autosomaalselt retsessiivsel viisil.
Kõige levinum tüüp, autosomaalselt dominantne pärandumine, esineb siis, kui muteerunud geenist on üks koopia (mida kannab üks vanematest). Hinnanguliselt on CMT edasikandumise tõenäosus igale sündinud lapsele 50%. [ 4 ]
Autosomaalselt retsessiivse pärandumise korral on haiguse tekkeks vaja kahte defektse geeni koopiat (üks igalt vanemalt, kellel haiguse tunnuseid ei esine).
40–50% juhtudest esineb autosomaalselt dominantne pärilik demüelinisatsioon ehk CMT tüüp 1; 12–26% juhtudest aksonaalne CMT ehk tüüp 2. Ja 10–15% juhtudest täheldatakse X-kromosoomiga seotud pärandumist. [ 5 ]
Sümptomid Charcot-Marie-Tooth'i haigus
Tavaliselt hakkavad selle haiguse esimesed nähud ilmnema lapsepõlves ja noorukieas ning arenevad järk-järgult kogu elu jooksul, kuigi sündroom võib endast märku anda ka hiljem. Sümptomite kombinatsioon on varieeruv ning haiguse progresseerumise kiirust ja raskusastet on võimatu ennustada.
Algstaadiumis esinevate tüüpiliste sümptomite hulka kuuluvad suurenenud üldine väsimus; jala-, pahkluude ja säärelihaste lihaste toonuse langus (nõrkus); reflekside puudumine. See raskendab jala liikumist ja viib düsbaasiani (kõnnihäire), mis avaldub jalgade kõrgemale tõusmises, sageli sagedaste komistamiste ja kukkumistega. Charcot-Marie-Toothi tõve tunnusteks väikelapsel võivad olla väljendunud kohmakus ja vanusele mitteomased kõndimisraskused, mis on seotud kahepoolse jala longusega. Iseloomulikud on ka jala deformatsioonid: kõrge jalavõlv (õõnes jalg) või tugev lamejalgsus, kõverad (haamrikujulised) varbad.
Lihashüpotoonia taustal varvastel kõndimise korral võib neuroloog kahtlustada, et lapsel on 4. tüüpi CMT, mille puhul lapsed ei pruugi noorukieas kõndida.
Haiguse progresseerumisel levib lihaste atroofia ja nõrkus ülajäsemetesse, mistõttu on raske peenmotoorikat ja tavalisi kätega seotud ülesandeid täita. Vähenenud puutetundlikkus ja võime tunda kuumust ja külma, samuti jalgade ja käte tuimus viitavad sensoorsete närvide aksonite kahjustusele.
Lapsepõlves avalduva Charcot-Marie-Toothi tõve 3. ja 6. tüübi korral täheldatakse sensoorset ataksiat (liigutuste ja tasakaalu koordinatsiooni häireid), lihaste tõmblemist ja värisemist, näonärvi kahjustust, nägemisnärvi atroofiat koos nüstagmusega ja kuulmislangust.
Hilisemates staadiumides võib esineda kontrollimatut värisemist (treemor) ja sagedasi lihaskrampe; liikumisprobleemid võivad põhjustada valu teket: lihas-, liigese- ja neuropaatilist.
Tüsistused ja tagajärjed
Charcot-Marie-Toothi haigusel võivad olla tüsistused ja tagajärjed, näiteks:
- sagedasemad nikastused ja luumurrud;
- periartikulaarsete lihaste ja kõõluste lühenemisega seotud kontraktuurid;
- skolioos (selgroo kõverus);
- hingamisprobleemid – kui diafragma lihaseid innerveerivad närvikiud on kahjustatud:
- iseseisva liikumise võime kaotus.
Diagnostika Charcot-Marie-Tooth'i haigus
Diagnoos hõlmab kliinilist läbivaatust, anamneesi (sh perekonnaanamneesi), neuroloogilist ja süsteemset läbivaatust.
Teste tehakse liikumisulatuse, tundlikkuse ja kõõluste reflekside kontrollimiseks. Närvijuhtivust saab hinnata instrumentaalse diagnostika abil – elektromüograafia või elektroneuromüograafia. Vajalikuks võib osutuda ka ultraheli või MRI. [ 6 ]
Geneetilised või DNA-testid vereproovis CMT-d põhjustavate kõige levinumate geneetiliste mutatsioonide tuvastamiseks on piiratud, kuna DNA-testid ei ole praegu kõigi haigustüüpide jaoks saadaval. Lisateabe saamiseks vaadake jaotist Geneetilised testid.
Mõnel juhul tehakse perifeerse närvi (tavaliselt suraalse närvi) biopsia.
Diferentseeritud diagnoos
Diferentsiaaldiagnoos hõlmab teisi perifeerseid neuropaatiaid, Duchenne'i lihasdüstroofiat, müelopaatilisi ja müasteenilisi sündroome, diabeetilist neuropaatiat, müelopaatiaid hulgiskleroosi ja amüotroofse lateraalskleroosi korral, Guillain-Barré sündroomi, peroneaalnärvi traumat ja selle atroofiat (sealhulgas nimmelülide vahele pigistamise korral), väikeaju või talamuse kahjustusi, samuti keemiaravi kõrvaltoimeid (tsütostaatikumidega, näiteks vinkristiin või paklitakseelravi ajal). [ 7 ]
Kellega ühendust võtta?
Ravi Charcot-Marie-Tooth'i haigus
Tänapäeval hõlmab selle päriliku haiguse ravi treeningteraapiat (mille eesmärk on lihaste tugevdamine ja venitamine); tegevusteraapiat (mis aitab käte lihasnõrkusega patsiente); ja ortopeediliste abivahendite kasutamist kõndimise hõlbustamiseks. Vajadusel määratakse valuvaigisteid või krambivastaseid ravimeid. [ 8 ]
Raske lamedate jalgade korral saab teha osteotoomiat ja kanna deformatsiooni korral on näidustatud kirurgiline korrektsioon – artrodees. [ 9 ]
Uuringud on käimas nii haiguse geneetilise komponendi kui ka ravimeetodite kohta. Tüvirakkude, teatud hormoonide, letsitiini või askorbiinhappe kasutamine ei ole veel positiivseid tulemusi andnud.
Kuid tänu hiljutistele uuringutele võib Charcot-Marie-Toothi tõve ravis lähitulevikus tõepoolest midagi uut ilmneda. Seega on Prantsuse ettevõte Pharnext alates 2014. aastast arendanud ja alates 2019. aasta keskpaigast on käimas kliinilised uuringud ravimiga PXT3003 CMT 1. tüübi raviks täiskasvanutel, mis pärsib PMP22 geeni suurenenud ekspressiooni, parandab perifeersete närvide müelinisatsiooni ja leevendab neuromuskulaarseid sümptomeid.
Meditsiinifirma Sarepta Therapeutics (USA) spetsialistid töötavad 1. tüüpi Charcot-Marie-Toothi tõve geeniteraapia loomise kallal. See teraapia kasutab kahjutut adeno-assotsieerunud viirust (AAV) perekonnast Dependovirus, millel on lineaarne üheahelaline DNA genoom, mis kannab organismi NTF3 geeni, mis kodeerib Schwanni närvirakkude toimimiseks vajalikku neurotrofiin-3 (NT-3) valku.
Helixmith alustab Lõuna-Koreas väljatöötatud geeniteraapia Engensis (VM202) kliinilisi uuringuid 2020. aasta lõpuks, et ravida 1. tüüpi CMT lihassümptomeid. [ 10 ]
Ärahoidmine
KMT ennetamine võib olla tulevaste vanemate geneetiline nõustamine, eriti kui kellelgi paarist on haiguse perekonnaajalugu. Siiski on tuvastatud de novo punktgeenimutatsioonide juhtumeid, st haiguse puudumisel perekonnaajaloos.
Raseduse ajal saab Charcot-Marie-Toothi haiguse tõenäosust tulevasel lapsel kontrollida koorionivilluse biopsiaga (10. kuni 13. rasedusnädalal) ja amnionivedeliku analüüsiga (15.-18. nädalal).
Prognoos
Üldiselt sõltub erinevat tüüpi Charcot-Marie-Toothi haiguse prognoos kliinilisest raskusastmest, kuid kõigil juhtudel progresseerub haigus aeglaselt. Paljudel patsientidel on puue, kuigi see ei vähenda oodatavat eluiga.