Artikli meditsiiniline ekspert

Uued väljaanded
Martin-Belli sündroom
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
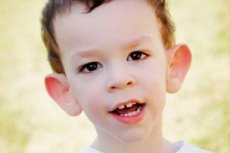
Martin-Belli sündroomi kirjeldasid arstid 1943. aastal ja selle järgi see ka nime sai. See haigus on geneetiline häire, mis seisneb vaimses alaarengus. 1969. aastal tuvastati sellele haigusele iseloomulikud muutused X-kromosoomis (distaalse käe haprus). 1991. aastal avastasid teadlased geeni, mis vastutab selle haiguse tekke eest. Seda haigust nimetatakse ka "hapra X sündroomiks". Nii poisid kui ka tüdrukud on haigusele vastuvõtlikud, kuid poisid haigestuvad sagedamini (3 korda).
Epidemioloogia
Martin-Belli sündroom on üsna levinud haigus: selle haiguse all kannatab 0,3–1,0 meest 1000-st ja 0,2–0,6 naist 1000-st. Lisaks sünnib Martin-Belli sündroomiga lapsi kõigil mandritel sama sagedusega. Ilmselgelt ei mõjuta rahvus, nahavärv, silmade kuju, elutingimused ja inimeste heaolu haiguse esinemist. Selle esinemissagedus on võrreldav ainult Downi sündroomi esinemissagedusega (1 haigus 600–800 vastsündinu kohta). Viiendik muudetud geeni meessoost kandjatest on terved, neil puuduvad kliinilised või geneetilised kõrvalekalded, ülejäänutel on vaimse alaarengu tunnused kergest kuni raskeni. Naissoost kandjatest on haiged veidi üle kolmandiku.
Fragiilse X-kromosoomi sündroom esineb ligikaudu ühel juhul 2500–4000 mehe ja ühel juhul 7000–8000 naise kohta. Kandluse levimus naiste seas on hinnanguliselt 1 juhtu 130–250-st; meeste seas on see hinnanguliselt 1 juhtu 250–800-st.
Põhjused Martin-Belli sündroom
Martin-Belli sündroom tekib organismi spetsiifilise valgu tootmise täieliku või osalise lakkamise tõttu. See tekib X-kromosoomis lokaliseeritud FMR1 geeni vastuse puudumise tõttu. Mutatsioon tekib geeni ümberstruktureerimise tagajärjel geeni seisundite (alleelide) ebastabiilsetest struktuurivariantidest, mitte algusest peale. Haigus kandub edasi ainult meesliini pidi ja mees ei pruugi tingimata haigestuda. Meessoost kandjad annavad geeni oma tütardele edasi muutumatul kujul, mistõttu nende vaimne alaareng ei ole ilmne. Geeni edasikandumisel emalt lastele geen muteerub ja ilmnevad kõik sellele haigusele iseloomulikud tunnused.
Pathogenesis
Martin-Belli sündroomi patogenees põhineb geeniaparaadi mutatsioonidel, mis viivad FMR-valgu tootmise blokeerimiseni. FMR-valk on organismile, eriti neuronites, elutähtis ja esineb erinevates kudedes. Uuringud näitavad, et FMR-valgud on otseselt seotud ajukoes toimuvate translatsioonide reguleerimise protsessidega. Selle valgu puudumine või selle piiratud tootmine organismis viib vaimse alaarenguni.
Haiguse patogeneesis peetakse geeni hüpermetülatsiooni võtmehäireks, kuid selle häire arengumehhanismi pole veel võimalik lõplikult kindlaks teha.
Samal ajal avastati ka patoloogia lookuse heterogeensus, mis on seotud polüallelismi ja polülokusega. Määrati kindlaks haiguse arengu alleelsete variantide olemasolu, mis on põhjustatud punktmutatsioonide olemasolust, samuti FMRL-tüüpi geeni hävimisest.
Patsientidel on ka kaks foolhappe suhtes tundlikku habrast kolmikut, mis paiknevad 300 kb kaugusel, samuti 1,5-2 miljonit bp kaugusel habrast kolmikust, mis sisaldab FMR1 geeni. FRAXE ja FRAXF geenides esinevate mutatsioonide mehhanism (need on tuvastatud eespool nimetatud habrastes kolmikutes) on seotud Martin Belli sündroomi häirete mehhanismiga. See mehhanism on tingitud GCC ja CGG korduste levikust, mis põhjustavad nn CpG saarte metüleerimist. Lisaks patoloogia klassikalisele vormile on ka kaks haruldast tüüpi, mis erinevad trinukleotiidide korduste laienemise tõttu (meeste ja naiste meioosi korral).
Leiti, et sündroomi klassikalises vormis puudub patsiendil spetsiaalne FMR1 tüüpi nukleotsütoplasmaatiline valk, mis täidab erinevate mRNA-de sidumise funktsiooni. Lisaks soodustab see valk kompleksi moodustumist, mis aitab ribosoomides translatsiooniprotsesse läbi viia.
Sümptomid Martin-Belli sündroom
Kuidas lastel haigust ära tunda? Millised on esimesed tunnused? Lapse elu esimestel kuudel on Martin-Belli sümptomit võimatu ära tunda, välja arvatud see, et mõnikord täheldatakse lihastoonuse langust. Aasta pärast on haiguse kliiniline pilt selgem: laps hakkab hilja kõndima ja rääkima, mõnikord puudub kõne täielikult. Ta on hüperaktiivne, vehib kätega juhuslikult, kardab rahvahulki ja müra, on kangekaelne, esineb teravaid vihapurskeid, emotsionaalset ebastabiilsust, tekivad epilepsiahood, ei loo silmsidet. Martin-Belli sündroomiga patsientidel reedab haigus ka välimuse järgi: kõrvad on punnis ja suured, otsaesine on raske, nägu on piklik, lõug on väljaulatuv, strabismus, laiad käed ja jalad. Neid iseloomustavad ka endokriinsed häired: sageli suur kaal, rasvumine, meestel suured munandid, varajane puberteet.

Martin-Belli sündroomiga patsientide intelligentsuse tase on väga erinev: kergest vaimsest alaarengust kuni raskete juhtudeni. Kui normaalse inimese intelligentsuskvoot (IQ) on keskmiselt 100 ja geeniusel 130, siis haigusele vastuvõtlikel inimestel on see 35–70.
Kõiki patoloogia kliinilisi sümptomeid saab iseloomustada peamiste ilmingute kolmikuga:
- oligofreenia (IQ on 35–50);
- düsmorfofoobia (täheldatakse väljaulatuvaid kõrvu ja prognatismi);
- makroorhidism, mis ilmneb pärast puberteedi algust.
Ligikaudu 80%-l patsientidest esineb ka bikuspidaalklapi prolaps.
Kuid sündroomi täielik vorm avaldub ainult 60% -l kõigist patsientidest. 10% -l tuvastatakse ainult vaimne alaareng ja ülejäänud patsientidel areneb haigus erineva sümptomite kombinatsiooniga.
Haiguse esimeste tunnuste hulgas, mis ilmnevad varases eas:
- haige lapsel on teiste eakaaslastega võrreldes märkimisväärne vaimne alaareng;
- tähelepanu ja keskendumishäired;
- tugev kangekaelsus;
- lapsed hakkavad üsna hilja kõndima ja rääkima;
- täheldatakse hüperaktiivsust ja kõne arenguhäireid;
- väga tugevad ja kontrollimatud vihahood;
- võib tekkida mutism - see on lapse täielik kõnevõime puudumine;
- beebi kogeb sotsiaalset ärevust ja on võimeline valju müra või muude valjude helide tõttu paanikasse sattuma;
- laps vehib kontrollimatult ja kaootiliselt kätega;
- täheldatakse häbelikkust, laps kardab rahvarohketes kohtades viibimist;
- mitmesuguste obsessiivsete ideede teke, ebastabiilne emotsionaalne seisund;
- Beebi võib olla vastumeelne inimestega silmsidet looma.
Täiskasvanutel täheldatakse järgmisi patoloogia sümptomeid:
- spetsiifiline välimus: piklik nägu raske laubaga, suured väljaulatuvad kõrvad, tugevalt väljaulatuv lõug;
- lamedad jalad, keskkõrvapõletik ja strabismus;
- puberteet algab üsna varakult;
- võib tekkida rasvumine;
- Martin-Belli sündroomi korral täheldatakse üsna sageli südamerikke;
- meestel täheldatakse munandite suurenemist;
- liigeste liigesed muutuvad väga liikuvaks;
- kaal ja pikkus suurenevad järsult.
Diagnostika Martin-Belli sündroom
Martin Belli sündroomi diagnoosimiseks peate pöörduma kvalifitseeritud geneetiku poole. Diagnoos pannakse pärast spetsiifilisi geneetilisi teste, mis võimaldavad teil tuvastada defektse kromosoomi.
Testid
Haiguse varases staadiumis kasutatakse tsütogeneetilist meetodit, mille käigus võetakse patsiendilt rakulise materjali fragment, millele lisatakse seejärel foolhapet, et esile kutsuda kromosoomides muutusi. Teatud aja möödudes tuvastatakse kromosoomi piirkond, kus on märgatav hõrenemine – see on märk fragiilse X-kromosoomi sündroomi olemasolust.
See test ei sobi aga haiguse hilisemates staadiumides diagnoosimiseks, sest selle täpsust vähendab foolhapet sisaldavate multivitamiinide laialdane kasutamine.
Martin-Belli sündroomi integreeritud diagnostika on molekulaargeneetiline uuring, mis seisneb geenis nn trinukleotiidide korduste arvu määramises.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentaalne diagnostika
Instrumentaalse diagnostika väga spetsiifiline meetod on PCR (polümeraasi ahelreaktsioon), mis võimaldab uurida X-kromosoomis sisalduvate aminohappejääkide struktuuri ja seeläbi määrata Martin Belli sündroomi olemasolu.
Samuti on olemas eraldi, veelgi spetsiifilisem patoloogia diagnostika meetod – PCR-i ja kapillaarelektroforeesi abil tuvastamise kombinatsioon. See meetod on väga täpne ja tuvastab kromosomaalset patoloogiat primaarse munasarjade puudulikkusega patsientidel, samuti ataksilise sündroomiga patsientidel.
Defekti olemasolu saab kindlaks teha pärast EEG diagnostika tegemist. Selle haigusega patsientidel on sarnane bioelektriline aju aktiivsus.
Millised testid on vajalikud?
Diferentseeritud diagnoos
Sündroomi kahtlustamiseks kasutatavad diferentseeritud meetodid hõlmavad järgmist:
- kliiniline - 97,5%-l patsientidest on ilmsed vaimse alaarengu tunnused (mõõdukas või sügav); 62%-l on väljaulatuvad suured kõrvad; 68,4%-l on suur väljaulatuv lõug ja otsmik; 68,4%-l poistest on suurenenud munandid, 41,4%-l on kõne iseärasused (ebaühtlane kõnetempo, kontrollimatu helitugevus jne);
- tsütogeenne - verd uuritakse lümfotsüütide kultuuri suhtes, määratakse kindlaks habras X-kromosoomiga rakkude arv 100 uuritud raku kohta;
- Elektroentsefalograafia - registreeritakse Martin-Belli sündroomile iseloomulikud aju elektriimpulsside muutused.
Kellega ühendust võtta?
Ravi Martin-Belli sündroom
Täiskasvanud patsientide ravis kasutatakse psühhostimulantidega antidepressante. Ravimiteraapia protsessi jälgib pidevalt psühholoog ja psühhiaater. Lisaks tehakse erakliinikutes mikroinjektsiooniprotseduure selliste ravimitega nagu Cerebrolysin (või selle derivaadid), samuti tsütomediinidega (näiteks Solcoseryl või Lidase).
Ataksia sündroomi tekkes kasutatakse verevedeldajaid ja nootroopikume. Lisaks on ette nähtud aminohapete segud ja angioprotektorid. Primaarse munasarjade puudulikkusega naistele on ette nähtud korrigeeriv ravi, kasutades taimseid ravimeid ja östrogeene.
Ravis kasutatakse ka glutamiini retseptori antagoniste.
Traditsiooniliselt hõlmab Martin-Belli sündroomi ravi ravimite kasutamist, mis mõjutavad haiguse sümptomeid, kuid mitte selle põhjust. See ravi hõlmab antidepressantide, neuroleptikumide ja psühhostimulantide määramist. Mitte kõik ravimid ei ole lastele näidustatud, seega on ravimite loetelu üsna piiratud. Neuroleptikumide hulka, mida saab kasutada pärast 3. eluaastat (kõige varasem väljakirjutamise vanus), kuuluvad haloperidool tilkade ja tablettidena, klorpromasiin lahuses ja peritsiasiin tilkadena. Seega arvutatakse haloperidooli annus lastele kehakaalu põhjal. Täiskasvanutele määratakse annus individuaalselt. Seda võetakse suu kaudu, alustades 0,5–5 mg-st 2–3 korda päevas, seejärel suurendatakse annust järk-järgult 10–15 mg-ni. Paranemise korral minnakse üle saavutatud seisundi säilitamiseks väiksemale annusele. Psühhomotoorse agitatsiooni korral määratakse 5–10 mg intramuskulaarselt või intravenoosselt, mitu korda on võimalik 30–40 minuti pärast. Päevane annus ei tohiks ületada 100 mg. Võimalikud on kõrvaltoimed iivelduse, oksendamise, lihasspasmide, suurenenud rõhu, arütmia jms kujul. Eakad inimesed peaksid võtma erilisi ettevaatusabinõusid, kuna on registreeritud äkilise südameseiskuse juhtumeid ja võib esineda tardiivset düskineesiat (tahtmatud liigutused).
Antidepressandid suurendavad aju struktuuride aktiivsust, leevendavad depressiooni, pinget ja parandavad meeleolu. Nende ravimite hulka, mida soovitatakse Martin-Belli sündroomi korral kasutada alates 5. eluaastast, kuuluvad klomipromiin, sertraliin, fluoksetiin ja fluvoksamiin. Seega võetakse fluoksetiini suu kaudu söögi ajal 1-2 korda (eelistatavalt päeva esimesel poolel), alustades 20 mg-st päevas, vajadusel suurendades seda 80 mg-ni. Eakatele ei ole soovitatav annus üle 60 mg. Ravikuuri määrab arst, kuid see ei tohi kesta kauem kui 5 nädalat.
Võimalikud kõrvaltoimed: pearinglus, ärevus, tinnitus, isutus, tahhükardia, tursed jne. Eakatele, südame-veresoonkonna haiguste ja diabeediga inimestele määramisel tuleb olla ettevaatlik.
Psühhostimulandid on psühhotroopsed ravimid, mida kasutatakse väliste stiimulite tajumise parandamiseks: need teravdavad kuulmist, reageerimisreaktsioone ja nägemist.
Diasepaami määratakse rahustina neurooside, ärevuse, epilepsiahoogude ja krampide korral. Seda võetakse suu kaudu, intravenoosselt, intramuskulaarselt, rektaalselt (pärasoole). See määratakse individuaalselt, olenevalt haiguse raskusastmest, väikseimad annused on 5-10 mg, päevas - 5-20 mg. Ravi kestus on 2-3 kuud. Lastele arvutatakse annus kehakaalu ja individuaalsete iseärasuste põhjal. Kõrvaltoimete hulka kuuluvad letargia, apaatia, unisus, iiveldus, kõhukinnisus. Ohtlik on seda kombineerida alkoholiga, võimalik on sõltuvus ravimist.
Martin-Belli sündroomi ravis on olnud juhtumeid seisundi paranemisest ja loomsetest materjalidest (ajust) valmistatud ravimite kasutuselevõtust: tserebrolüsaat, tserebrolüsiin, tserebrolüsaat-M. Nende ravimite peamised komponendid on peptiidid, mis soodustavad valgu tootmist neuronites, täiendades seeläbi puuduvat valku. Tserebrolüsiini manustatakse 5-10 ml joana, ravikuur koosneb 20-30 süstist. Ravimit määratakse lastele alates üheaastasest eluaastast, manustades intramuskulaarselt iga päev 1-2 ml kuu jooksul. Võimalikud on korduvad manustamissessioonid. Kõrvaltoimed palaviku näol, vastunäidustatud rasedatele.
Haigust püüti ravida foolhappega, kuid paranes ainult käitumuslik aspekt (vähenes agressiivsuse ja hüperaktiivsuse tase, paranes kõne) ning intellektuaalsel tasandil ei muutunud midagi. Haiguse seisundi parandamiseks määratakse foolhape, näidustatud on füsioteraapia meetodid, logopeediline ravi, pedagoogiline ja sotsiaalne korrektsioon.
Liitiumipreparaate peetakse samuti efektiivseks, kuna need aitavad parandada patsiendi kohanemist sotsiaalse keskkonnaga ja kognitiivset aktiivsust. Lisaks reguleerivad nad ka tema käitumist ühiskonnas.
Martin-Belli sündroomi korral on võimalik antidepressantidena kasutada ravimtaimi. Pingeid ja ärevust leevendavate ning und parandavate ravimtaimede hulka kuuluvad palderjan, piparmünt, tüümian, naistepuna ja kummel. Keedised valmistatakse järgmiselt: 1 teelusikatäie kuivatatud ürtide kohta on vaja klaasi keeva vett, keediseid lastakse tõmmata vähemalt 20 minutit, võttes neid peamiselt õhtul enne magamaminekut või pärastlõunal. Heaks lisandiks oleks lusikatäis mett.
Füsioteraapia
Neuroloogiliste ilmingute kõrvaldamiseks viiakse läbi spetsiaalseid füsioterapeutilisi protseduure - näiteks basseiniharjutused, lihaste lõdvestamine ja nõelravi.
Kirurgiline ravi
Oluliseks ravietapiks peetakse ka plastilise kirurgia meetodeid - operatsioone, mis aitavad parandada patsiendi välimust. Tehakse jäsemete ja kõrvakeste, samuti suguelundite plastiline kirurgia. Samuti tehakse günekomastia korrigeerimist koos epispadiatega, samuti muid välimuse defekte.
Ärahoidmine
Ainus haiguse ennetamise meetod on rasedate sünnieelne skriining. On olemas spetsiaalsed uuringud, mis võimaldavad patoloogiat varakult avastada, mille järel on soovitatav rasedus katkestada. Alternatiivina kasutatakse IVF-i, mis aitab lapsel pärida terve X-kromosoomi.
Patsiendi ennetamine sõltub sellest, kas geenimutatsioon on uuesti tekkinud või päritud. Selleks viiakse läbi molekulaargeneetiline diagnostika. Asjaolu, et test ei näidanud sugulastel "habrast X-kromosoomi", räägib mutatsiooni "värskuse" kasuks, mis tähendab, et Martin-Belli sündroomiga lapse saamise risk on väga väike. Peredes, kus on haigeid inimesi, aitab test vältida korduvaid juhtumeid.
Prognoos
Martin-Belli sündroomi prognoos on eluks ajaks soodne, kuid mitte paranemiseks. Oodatav eluiga sõltub haiguse raskusastmest ja sellega kaasnevatest defektidest. Patsient saab elada normaalset elu. Martin-Belli sündroomi raskete vormide korral on patsientidel eluaegse puude oht.
Oodatav eluiga
Martin Belli sündroomil ei ole tervisele tõsist negatiivset mõju, seega enamiku selle patoloogiaga diagnoositud inimeste eluiga ei erine standardnäitajatest.