Artikli meditsiiniline ekspert

Uued väljaanded
Dandy-Walkeri sündroom
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
Dandy-Walkeri sündroom on haruldane ja üsna raske väikeaju ja neljanda vatsakese väärareng. Seda peetakse lastel kaasasündinud anomaaliaks ja see esineb sageli sagedusega 1:25 000. Seda defekti iseloomustab väikeaju vermise alaareng või hüpoplaasia, neljanda vatsakese tsüstiline laienemine ja tagumise koljulohu laienemine. Sündroomiga võivad kaasneda mitmed teiste organite anomaaliad, kuid peamised neist on kolm eespool loetletud. Ligikaudu 70–90% patsientidest esineb hüdrotsefaalia, mis sageli tekib vahetult pärast sündi. Õnneks saab patoloogiat avastada isegi raseduse ajal.
Põhjused Dandy-Walkeri sündroom
Kahjuks ei suuda isegi tänapäeva meditsiin veel kindlalt vastata küsimusele, mis põhjustab Dandy-Walkeri sündroomi teket. Loomulikult on olemas mõned riskifaktorid, mis ühel või teisel juhul võivad viia emakasisesete defektide tekkeni. Selle anomaalia kujunemisel mängivad olulist rolli järgmised tegurid:
- Viirushaigused, mida ema raseduse ajal põeb (eriti tuleb olla ettevaatlik infektsioonide suhtes esimese kolme kuu jooksul).
- Tsütomegaloviiruse infektsioon.
- Punetised.
- Suhkurtõbi või muud haigused, mis on seotud ainevahetushäiretega rasedal emal.
Üsna sageli esinevad alkoholismi või narkosõltuvuse all kannatavatel naistel emakasisese arengu ajal mitmesugused loote patoloogiad. Enamasti sünnitavad just sellised emad Dandy-Walkeri sündroomiga lapsi. Kui arst ultraheli ajal näeb, et loode areneb ebanormaalselt, soovitab ta raseduse katkestada. Väga harvadel juhtudel tekib Dandy-Walkeri sündroom pärilikel põhjustel.
Pathogenesis
Dandy-Walkeri sündroomi kirjeldasid esmakordselt Dandy ja Blackfan 1914. aastal. Pärast esialgset kirjeldust on läbi viidud täiendavaid uuringuid, mis on kirjeldanud selle defekti erinevaid morfoloogilisi tunnuseid.
D'Agostino 1963. aasta ja Harti 1972. aasta uuringud tuvastasid Dandy-Walkeri sündroomi iseloomuliku triaadi, nimelt:
- Ussi täielik või osaline agenees
- Neljanda vatsakese tsüstiline dilatatsioon
- Tagumise koljuõõne suurenemine koos külgmiste paranasaalsete siinuste ja väikeaju ülespoole nihkumisega.
See triaad on tavaliselt seotud supratentoriaalse hüdrotsefaaliaga, mis on tüsistused ja ei ole osa väärarengukompleksist.
Klassikaliselt jagunevad tagumise lohu tsüstilised väärarengud Dandy-Walkeri sündroomiks, Dandy-Walkeri variandiks, mega cisterna magnaks ja tagumise lohu arahnoidaaltsüstiks - need väärarengud moodustavad Dandy-Walkeri kompleksi.
Dandy-Walkeri variant ühendab vermise hüpoplaasia ja neljanda vatsakese tsüstilise dilatatsiooni ilma tagumise lohu suurenemiseta.
See sündroom on kaasasündinud patoloogia, mis väljendub selles, et tserebrospinaalvedeliku rajad ja väikeaju ei ole täielikult välja arenenud. Enamasti puudutab anomaalia avasid, mis ühendavad kahte vatsakest (kolmandat ja neljandat) aju suure tsisterniga, ja neid, mis ühendavad samu vatsakesi ajukelme subarahnoidaalse ruumiga. Tserebrospinaalvedeliku väljavoolu häire tõttu tekib tagumises koljulohus tserebrospinaalvedeliku tsüstiline moodustis ja neljanda vatsakese sekundaarse tsüstilise laienemise tagajärjel obstruktiivne hüdrotsefaalia (70–90% juhtudest).
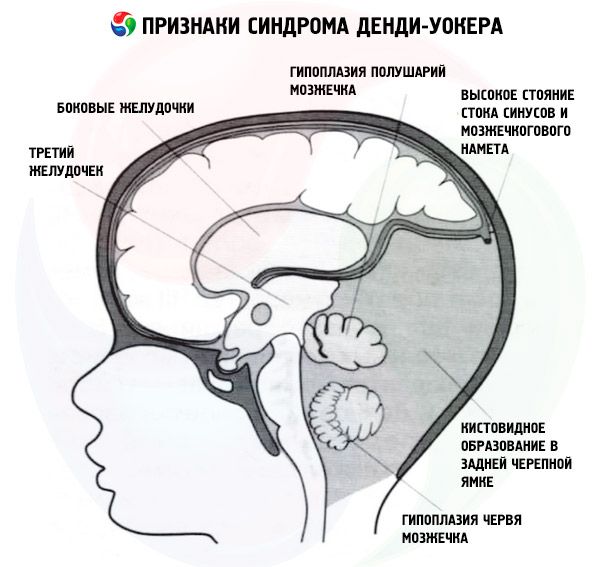
Dandy-Walkeri anomaaliaga seotud kesknärvisüsteemi väärarengud (70% juhtudest):
- Mõjukeha düsgenees (20-25%).
- Corpus callosumi lipoom.
- Holoprosentsefaalia (25%).
- Porentsefaalia.
- Tsingulaatdüsplaasia (25%).
- Skisentsefaalia.
- Polümikrogüüria/halli aine heterotoopia (5–10%).
- Väikeaju heterotoopia.
- Kuklaluu entsefalocele (7%).
- Mikrotsefaalia.
- Dermoidne tsüst.
- Väikeaju lobulite väärarengud (25%).
- Alumise olivaartuuma väärarengud.
- Hamartoma hallis mugulas.
- Süringomüelia.
- Klippel-Feili deformatsioon.
- Seljaajusong.
Muud haruldased seotud KNS-i väärarengud (20–33% juhtudest):
- Suuõõne ja suulae deformatsioonid (6%).
- Polüdaktüülia ja sündaktüülia.
- Südame arengu anomaaliad.
- Kuseteede häired (polütsüstiline neeruhaigus).
- Võrkkesta katarakt, koroidaalne düsgenees, koloboom.
- Hemangioom näol.
- Hüpertelorism.
- Meckel-Gruberi sündroom.
- Neurokutaanne melanoos.
Sümptomid Dandy-Walkeri sündroom
Seda lapse arengu kõrvalekallet saab ultraheli abil näha juba raseduse esimestel kuudel. Eelkõige on kõik selle haiguse sümptomid nähtavad juba 20. nädalal. Patoloogia arengu peamised tõendid on mitmesugused ajukahjustuse tunnused: koljuõõnes olevad tsüstid on selgelt nähtavad, väikeaju on halvasti arenenud, neljas vatsake on liiga laienenud. Samal ajal muutuvad ultraheli tunnused loote arenedes selgemaks.
Aja jooksul tekivad kõvas suulaes ja huules lõhed, neerude ebanormaalne areng ja sündaktüülia. Kui mingil põhjusel raseduse ajal ultraheli ei tehtud, muutuvad haiguse sümptomid selgelt nähtavaks kohe pärast lapse sündi. Koljusisene kõrge rõhk on sellised lapsed väga rahutud. Neil tekib hüdrotsefaalia koos lihasspasmide ja nüstagmusega. Mõnel juhul ei pruugi hüdrotsefaaliale mingeid märke olla.
Esimesed märgid
- Magendie ja Luschka ava atresia (sulgemine või kaasasündinud puudumine).
- Koljuõõne tagumine osa suureneb.
- Väikeaju poolkerade atroofia.
- Ilmuvad tsüstilised moodustised, sealhulgas fistulid.
- Erineva astmega hüdrotsefaalia.
Kõik need esimesed märgid on loote ultraheli ajal selgelt nähtavad. Seetõttu on rasedate naiste jaoks nii oluline läbida kõik testid ja uuringud õigeaegselt.
Dandy-Walkeri sündroom lastel
A priori peetakse Dandy-Walkeri sündroomi lastehaiguseks. Õnneks on see üsna haruldane (ainult üks juhtum 25 000 vastsündinu kohta). Selle sümptomeid võib näha juba emakas, kuid kui mingil põhjusel seda ei tehtud, siis tekivad selle haigusega lastel vanusega üsna tugevad ja rasked tüsistused.
Aja jooksul ilmnevad märgatavad väikeaju sümptomid. Vanematel lastel hakkab liigutuste koordinatsioon halvenema, mistõttu on neil üsna raske liikuda ja kõndida (mõnikord isegi võimatu). Peamine sümptom on raske vaimne alaareng, mida on peaaegu võimatu ravida. Ilmnevad ka kaasnevad probleemid: neeruhaigus, südamehaigus, sõrmede, näo, käte ebanormaalne areng, halb nägemine.
 [ 16 ]
[ 16 ]
Dandy-Walkeri sündroom täiskasvanutel
Dandy-Walkeri sündroom on anatoomiline ja kliiniline variant pahaloomulisest hüdrotsefaaliast. Seda haigust iseloomustab aju avade atresia ja vatsakeste (kolmanda ja neljanda) laienemine.
Hoolimata asjaolust, et seda patoloogiat peetakse kaasasündinud, hakkavad selle esimesed märgid väga harvadel juhtudel ilmnema varases lapsepõlves (nelja-aastaselt) või isegi hiljem. Pikka aega ei ole sellel kaasasündinud defektil mingeid sümptomeid. Dekompensatsioon tekib mõnikord ainult kooliealistel lastel või noorukitel. Erandjuhtudel ilmnevad Dandy-Walkeri sündroomi esimesed sümptomid täiskasvanutel.
Täiskasvanute haiguse peamiste sümptomite hulgas on järgmised:
- Pea suurus suureneb järk-järgult.
- Pea tagaosa luud hakkavad välja paistma: need justkui punnitavad välja.
- Liigutuste koordineerimine on tugevalt häiritud, ilmnevad ebaselged ja laiaulatuslikud liigutused.
- Tekib nüstagmus, mis avaldub silmade võnkuva liikumisena küljelt küljele.
- Sagedased krambid koos krampidega.
- Lihastoonus suureneb oluliselt (mõnikord kuni spastilisuseni). Lihased on pidevas pinges.
- Vaimse alaarengu areng, mis avaldub selles, et inimene ei tunne sugulasi ära, tal on raskusi tähtede lugemise ja eristamisega ning ta ei oska kirjutada.
[ 17 ], [ 18 ], [ 19 ], [ 20 ]
Dandy-Walkeri sündroom raseduse ajal
Tavaliselt on Dandy-Walkeri sündroomi ultraheli abil näha juba 18-20 rasedusnädalal. Sel perioodil võib olla märgatav väikeajuussi täielik või osaline agenees. Väga harvadel juhtudel on selle haiguse sünnieelne diagnoosimine võimalik varasemas staadiumis.
Tüsistused ja tagajärjed
Muidugi on paljud kaasasündinud patoloogiad väga tõsised, kuid Dandy-Walkeri sündroomi peetakse laste üheks kõige raskemaks anomaaliaks. See haigus toob kaasa üsna keerulisi tagajärgi:
- Vaimne puue jääb kogu eluks.
- Sageli jääb alles ka neuroloogiline defekt: lihastoonus on suurenenud ja liigutuste koordineerimine on vale.
- Enamikul juhtudel võib eeldada surmavat tulemust.
Diagnostika Dandy-Walkeri sündroom
Esiteks diagnoosib arst kaebused ja haigusloo (kui Dandy-Walkeri sündroom ilmneb pärast sündi). See etapp hõlmab vastuseid mõnele küsimusele:
- Kas teie peres on midagi sellist varem juhtunud?
- Millises vanuses hakkasid ilmnema esimesed haiguse tunnused?
- Millal hakkas liigutuste koordinatsioon häiruma, pea kasvama ja lihastoonus muutuma?
Järgmisena peaks spetsialist läbi viima neuroloogilise läbivaatuse. Selles etapis võib täheldada nüstagmuse teket (silmade tahtmatud võnkuvad liigutused erinevates suundades), fontanelli punnitust, lihastoonuse langust ja pea suuruse suurenemist.
Ultraheliuuringu läbiviimine raseduse ajal aitab näha sündroomi esimesi märke lootel. Lisaks saab sama meetodit kasutada haiguse hilisemates staadiumides südame defektide kontrollimiseks.
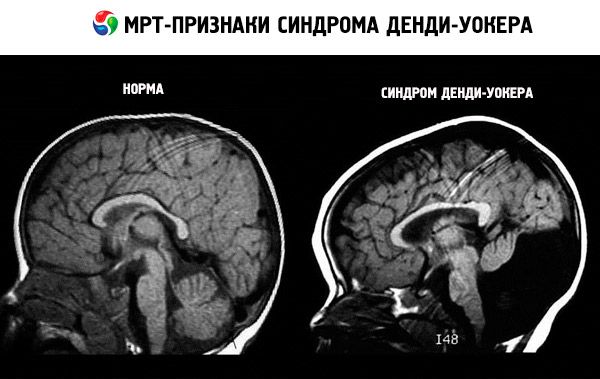
Aju magnetresonantstomograafia (MRI) abil on võimalik näha pea suuruse suurenemise astet, tsüsti esinemist tagumises koljuõõnes, vatsakese suurenemist, väikeaju ebanormaalset arengut ja hüdrotsefaaliat.
Mõnel juhul on vaja konsulteerida neurokirurgi või meditsiinigeneetikuga.
Instrumentaalne diagnostika
Dandy-Walkeri sündroomi esimeste sümptomite avastamiseks on instrumentaalse diagnostika kõige levinumad meetodid ultraheli (loote ultraheliuuring) ja MRI (magnetresonantstomograafia). Reeglina on lapse ebanormaalne areng märgatav esimese ultraheli ajal, mis tehakse 18-20 rasedusnädalal. Kui spetsialist ei suuda täpset diagnoosi panna või on vaja selle kinnitamist, võib ta määrata MRI. MRI abil tehtud diagnoos on 99% täpne.
Kuidas uurida?
Millised testid on vajalikud?
Diferentseeritud diagnoos
Dandy-Walkeri sündroomi diferentsiaaldiagnostikat tehakse tavaliselt aju suure tsisterni laienemise korral, mis tekib väikeaju põletikulise või toksilise hüpoplaasia, samuti retrotserebraalse tsüstiga. Kuid siin tasub arvestada, et Dandy-Walkeri sündroomi korral on väikeaju vermikul patognomooniline defekt, mida teiste hüpoplaasiate korral ei täheldata. Selle patoloogia diferentsiaaldiagnostika läbiviimiseks arahnoidaalsete tsüstide korral on vaja teha magnetresonantstomograafia (MRI).
Kellega ühendust võtta?
Ravi Dandy-Walkeri sündroom
Dandy-Walkeri sündroomil on erinev raskusaste. Reeglina surevad selle anomaalia raske vormiga sündinud lapsed esimesel elukuul. Kui defekt on eluga kooskõlas, peetakse lapse vaimset arengut väga problemaatiliseks. Vanemad peaksid olema valmis selleks, et lapsel on väga madal intelligentsus, mida ei saa tugipedagoogika abil muuta. Dandy-Walkeri sündroomiga lapse arengu prognoos on äärmiselt ebasoodne.
Sellise anomaalia ravimisel pole praktiliselt mingit mõtet. Aga kui hüdrotsefaalia hakkab suurenema, on vaja kirurgilist sekkumist. Koljusisest rõhku saab vähendada aju neljanda vatsakese šunteerimisega.
Dandy-Walkeri sündroomi ravi saab olla ainult sümptomaatiline. Lapse vaimset alaarengut ei saa korrigeerida, keha motoorsed funktsioonid on tugevalt häiritud, mistõttu sellised lapsed ei saa tavaliselt iseseisvalt istuda ega seista.
Ravimisel on väga oluline meeles pidada kaasnevaid südame-, neeru- ja näo-lõualuu patoloogia haigusi. Need vajavad sageli kvalifitseeritud meditsiinilist sekkumist.
Kui arst tuvastab lootel selle anomaalia, peavad vanemad tegema üsna raske valiku: katkestada rasedus või sünnitada laps, kes kannatab kogu ülejäänud elu ravimatu patoloogia all.
Kirurgiline ravi
Dandy-Walkeri sündroomi ainus kirurgiline ravi on šunteerimismeetod, mis aitab vähendada hüdrotsefaaliasümptomite raskust ja parandada tserebrospinaalvedeliku väljavoolu.
Ärahoidmine
Kuna Dandy-Walkeri sündroom on pärilik haigus, peetakse selle ennetamist mõttetuks. Raseduse ajal on soovitatav regulaarselt külastada günekoloogi-ämmaemandat (esimesel trimestril üks kord kuus, teisel trimestril iga kahe kuni kolme nädala tagant ja kolmandal trimestril iga seitsme kuni kümne päeva tagant). Püüdke õigeaegselt (enne 12. rasedusnädalat) naistekliinikusse registreeruda.
Prognoos
Dandy-Walkeri sündroomi prognoos on katastroofiliselt ebasoodne. 50% juhtudest sureb laps esimesel eluaastal. Need lapsed, kes ellu jäävad, peavad olema pidevalt arstide valvsa silma all. Neil on äärmiselt madal intelligentsuse tase, mida ei saa korrigeerida. Sündroom esineb väljendunud funktsionaalse neuroloogilise seisundi korral.
[ 39 ]
Puue
Kesknärvisüsteemi kaasasündinud defektid, eriti Dandy-Walkeri sündroom, põhjustavad esimestest eluminutitest alates rasket puuet.