Artikli meditsiiniline ekspert

Uued väljaanded
Keratoderma: põhjused, sümptomid, diagnoosimine, ravi
Viimati vaadatud: 07.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
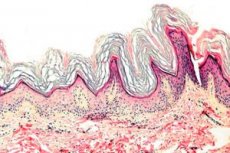
Keratoderma on dermatooside rühm, mida iseloomustab keratiniseerumisprotsessi häire - liigne sarvkoe moodustumine peamiselt peopesadel ja jalataldadel.
Haiguse põhjuseid ja patogeneesi pole täielikult selgitatud. Uuringud on kindlaks teinud, et keratodermiad tekivad keratiini 6, 9, 16 kodeerivate geenide mutatsioonide tagajärjel. Patogeneesis on suur tähtsus A-vitamiini puudusel, hormonaalsetel häiretel, peamiselt sugunäärmete talitlushäiretel, bakteriaalsetel ja viirusnakkustel. Need on üks pärilike haiguste ja siseorganite kasvajate (parapsoriaatilise keratodermia) sümptomitest.
Sümptomid. Eristatakse difuusset (Unna-Tosti keratoderma, Meleda keratoderma, Papillon-Lefevre keratoderma, moonutav keratoderma ja sündroomid, mille üheks peamiseks sümptomiks on difuusne keratoderma) ja fokaalset (dissemineerunud täpiline Fischer-Buschke keratoderma, Kosti akrokeratoelastoidoos, Bruhauer-Franzesthesti piiratud keratoderma, Fuchsi lineaarne keratoderma jne) keratodermat.
Winy-Tosti keratooderma (sünonüümid: peopesade ja jalataldade kaasasündinud iktüoos, Winy-Tosti sündroom) kandub edasi autosomaalselt dominantsel viisil. Peopesade ja jalataldade (mõnikord ainult jalataldade) nahal esineb difuusne liigne keratiniseerumine, mis tekib kahel esimesel eluaastal. Naha patoloogiline protsess algab peopesade ja jalataldade naha kerge paksenemisega kahvatu erüteemi riba kujul terve naha piiril. Aja jooksul ilmuvad nende pinnale siledad, kollakad sarvkihid. Kahjustus levib harva randmete või sõrmede seljale. Mõnedel patsientidel võivad tekkida pindmised või sügavad praod ja täheldatakse lokaalset hüperhidroosi. Autori vaadeldud patsiendil, emapoolsel onul, vennal ja pojal, esines Winy-Tosti keratoodermat.
Kirjeldatakse küünte (paksenemise), hammaste ja juuste kahjustuse juhtumeid Winy-Tosti keratoderma korral koos erinevate skeleti anomaaliate ja siseorganite, närvisüsteemi ja endokriinsüsteemi patoloogiatega.
Histopatoloogia. Histoloogiline uuring näitab märgatavat hüperkeratoosi, granuloosi, akantoosi ja väikeseid põletikulisi infiltraate dermise ülemises kihis. Diferentsiaaldiagnoos. Haigus tuleb eristada teist tüüpi keratodermiast.
Meleda keratooderma (sünonüümid: Meleda tõbi, kaasasündinud progresseeruv akrokeratoom, Siemensi palmoplantaarne transgradientset keratoos, Kogoy pärilik palmoplantaarne progresseeruv keratoos) pärandub autosomaalselt retsessiivselt. Seda keratooderma vormi iseloomustavad paksud, kollakaspruunid sarvkihid sügavate pragudega. Kahjustuse servades on nähtav mitme millimeetri laiune violetne-lilla äär. Protsess levib tavaliselt käte ja jalgade tagaküljele, käsivartele ja säärtele. Enamikul patsientidest esineb lokaalset hüperhidroosi. Sellega seoses muutub peopesade ja jalataldade pind kergelt niiskeks ja kaetuks mustade täppidega (higinäärmete kanalid).
Haigus võib areneda 15–20-aastaselt. Küüned paksenevad ja deformeeruvad.
Histopatoloogia. Histoloogiline uuring näitab hüperkeratoosi, mõnikord akantoosi ja kroonilist põletikulist infiltraati papillaarses dermises.
Diferentsiaaldiagnoos. Melela keratodermat tuleb eristada Unna-Tosti keratodermast.
Keratoderma Papillon-Lefevre (sünonüüm: palmoplantaarne hüperkeratoos koos periodontiidiga) pärandub autosomaalselt retsessiivselt.
Haigus avaldub 2.-3. eluaastal. Haiguse kliiniline pilt on sarnane Melela tõvega. Lisaks on iseloomulikud muutused hammastes (piima- ja jäävhammaste tuleku kõrvalekalded koos kaariese tekkega, gingiviit, kiiresti progresseeruv parodontoos koos enneaegse hammaste väljalangemisega).
Histopatoloogia. Histoloogiline uuring näitab epidermise kõigi kihtide, eriti sarvkihi paksenemist ning dermises lümfotsüütide ja histiotsüütide ebaolulisi rakulisi klastreid.
Diferentsiaaldiagnoos. Haigust tuleks eristada teistest keratodermiatest. Oluline eristav tunnus on iseloomulik hambapatoloogia, mida ei esine teistes päriliku difuusse keratodermia vormides.
Keratoderma mutilans (sünonüümid: Fonwinkeli sündroom, pärilik mutileeriv keratoom) on autosomaalselt dominantselt päranduv difuusne keratodermia tüüp. See tekib teisel eluaastal ja seda iseloomustavad difuussed sarvjas ladestused peopesade ja jalataldade nahal koos hüperhidroosiga. Aja jooksul tekivad sõrmedele nööritaolised sooned, mis viivad kontraktuuride ja sõrmede spontaanse amputatsioonini. Follikulaarne keratoos avaldub käeseljal, samuti küünarnuki- ja põlveliigeste piirkonnas. Küüneplaadid muutuvad (sageli nagu kellaklaasid). On kirjeldatud hüpogonadismi, rubiinkarva alopeetsia, kuulmislanguse ja pahüonühhia juhtumeid.
Histopatoloogia. Histoloogiline uuring näitab rasket hüperkeratoosi, granuloosi, akantoosi ja väikeseid põletikulisi infiltraate dermises, mis koosnevad lümfotsüütidest ja histiotsüütidest.
Diferentsiaaldiagnostika. Mutiliseeriva keratooderma eristamisel teistest difuusse keratooderma vormidest tuleks kõigepealt arvesse võtta moonutusefekti, mis ei ole teistele vormidele tüüpiline. Kõikide difuusse keratooderma vormide diferentsiaaldiagnostika läbiviimisel tuleb meeles pidada, et see võib olla mitmete pärilike sündroomide üks peamisi sümptomeid.
Ravi. Neotigazone on näidustatud keratodermia üldises ravis. Ravimi annus sõltub protsessi raskusastmest ja on 0,3–1 mg/kg patsiendi kehakaalu kohta. Neotigazone'i puudumisel on soovitatav A-vitamiini manustada annuses 100–300 000 mg päevas pikka aega. Väline ravi seisneb aromaatsete retinoidide, keratolüütiliste ja steroidsete ainetega salvide kasutamises.
 [
[ Mis teid häirib?
Mida tuleb uurida?
Kuidas uurida?