Artikli meditsiiniline ekspert

Uued väljaanded
Pärilik nefriit (Alporti sündroom) lastel
Viimati vaadatud: 05.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
Pärilik nefriit (Alporti sündroom) on geneetiliselt määratud pärilik mitteimmuunne glomerulopaatia, mis avaldub hematuurias (mõnikord koos proteinuuriaga), neerufunktsiooni progresseeruvas languses koos kroonilise neerupuudulikkuse tekkega, sageli koos sensoorse kurtuse ja nägemiskahjustusega.
Haigust kirjeldas esmakordselt 1902. aastal L. G. Guthrie, kes jälgis perekonda, kus hematuuriat täheldati mitme põlvkonna jooksul. 1915. aastal kirjeldas A. F. Hurst ureemia teket sama perekonna liikmetel. 1927. aastal tuvastas A. Alport esmakordselt kuulmislanguse mitmel hematuuriaga sugulasel. 1950. aastatel kirjeldati sarnase haiguse korral esinevaid silmakahjustusi. 1972. aastal avastasid Hinglais jt neerukoe morfoloogilise uuringu käigus päriliku hematuuriaga patsientidel glomerulaarsete basaalmembraanide ebaühtlase laienemise ja kihistumise. 1985. aastal tuvastati päriliku nefriidi geneetiline alus - mutatsioon IV tüüpi kollageeni geenis (Fiengold jt, 1985).
Haiguse geneetilise olemuse uurimine võimaldas meil järeldada, et päriliku nefriidi fenotüüpiliste ilmingute erinevused (kuulmislangusega või ilma) tulenevad mutantse geeni ekspressiooniastmest. Seega peetakse praegu kõiki kliinilisi variante ühe haiguse ilminguteks ja termin "pärilik nefriit" on sünonüümne terminiga "Alporti sündroom".
Epidemioloogiliste uuringute kohaselt esineb pärilikku nefriiti sagedusega 17 juhtu 100 000 lapse kohta.
 [
[ Alporti sündroomi põhjused
Haiguse geneetiline alus on mutatsioon IV tüüpi kollageeni α-5 ahela geenis. See tüüp on universaalne neeru basaalmembraanidele, kohleaaraparaadile, läätsekapslile, võrkkestale ja silma sarvkestale, mida on tõestatud uuringutes, milles kasutati selle kollageenifraktsiooni vastaseid monoklonaalseid antikehi. Hiljuti on näidatud võimalust kasutada DNA-sonde päriliku nefriidi sünnieelseks diagnostikaks.
Rõhutatakse kõigi pereliikmete DNA-sondidega testimise olulisust mutantse geeni kandjate tuvastamiseks, millel on suur tähtsus selle haigusega perede meditsiinilise ja geneetilise nõustamise läbiviimisel. Siiski ei ole kuni 20% peredest neeruhaigusega sugulasi, mis viitab ebanormaalse geeni spontaansete mutatsioonide kõrgele sagedusele. Enamikul päriliku nefriidiga patsientidest on peredes neeruhaiguse, kuulmislanguse ja nägemispatoloogiaga isikuid; olulised on veresugulusabielud inimeste vahel, kellel on üks või mitu esivanemat, kuna sugulaste abielus suureneb tõenäosus saada samu geene mõlemalt vanemalt. On kindlaks tehtud autosomaalselt dominantne, autosomaalselt retsessiivne ja dominantne X-kromosoomiga seotud ülekandetee.
Lastel eristatakse kõige sagedamini kolme tüüpi pärilikku nefriiti: Alporti sündroom, pärilik nefriit ilma kuulmislanguseta ja perekondlik healoomuline hematuuria.
Alporti sündroom on pärilik nefriit, millega kaasneb kuulmislangus. See põhineb neerude, kõrva ja silma struktuuride glomerulaarse basaalmembraani kollageeni struktuuri kombineeritud defektil. Klassikalise Alporti sündroomi geen asub X-kromosoomi pika haru lookuses 21-22q. Enamasti pärandub see dominantselt, seotuna X-kromosoomiga. Sellega seoses on Alporti sündroom meestel raskem, kuna naistel kompenseerib mutantse geeni funktsiooni teise, kahjustamata kromosoomi terve alleel.
Päriliku nefriidi tekke geneetiliseks aluseks on IV tüüpi kollageeni alfa-ahelate geenide mutatsioonid. IV tüüpi kollageen G-st on teada kuus alfa-ahelat: a5- ja a6-ahelate geenid (Col4A5 ja Col4A5) paiknevad X-kromosoomi pikal harul 21-22q tsoonis; a3- ja a4-ahelate geenid (Col4A3 ja Col4A4) asuvad 2. kromosoomis; a1- ja a2-ahelate geenid (Col4A1 ja Col4A2) asuvad 13. kromosoomis.
Enamikul juhtudel (80–85%) tuvastatakse haiguse X-kromosoomiga seotud pärimismuster, mis on seotud Col4A5 geeni kahjustusega deletsiooni, punktmutatsioonide või splaissinguhäirete tagajärjel. Praegu on leitud üle 200 Col4A5 geeni mutatsiooni, mis vastutavad IV tüüpi kollageeni a5-ahelate sünteesi häire eest. Seda tüüpi pärilikkuse korral avaldub haigus mõlema soo lastel, kuid poistel on see raskem.
IV tüüpi kollageeni a3 ja a4 ahelate sünteesi eest vastutavate geenide Col4A3 ja Col4A4 lookuste mutatsioonid päranduvad autosomaalselt. Uuringute kohaselt on autosomaalselt dominantne pärandumistüüp täheldatud 16% päriliku nefriidi juhtudest ja autosomaalselt retsessiivne tüüp 6% patsientidest. Col4A3 ja Col4A4 geenide mutatsioonide kohta on teada umbes 10 varianti.
Mutatsioonide tagajärjeks on IV tüüpi kollageeni koostiseprotsesside rikkumine, mis viib selle struktuuri rikkumiseni. IV tüüpi kollageen on üks glomerulaarse basaalmembraani, kohleaaparaadi ja silmaläätse peamisi komponente, mille patoloogiat avastatakse päriliku nefriidi kliinikus.
IV tüüpi kollageen, mis on osa glomerulaarsest basaalmembraanist, koosneb peamiselt kahest a1-ahelast (IV) ja ühest a2-ahelast (IV) ning sisaldab ka a3-, a4- ja a5-ahelaid. Kõige sagedamini kaasneb X-seotud pärandumise korral Col4A5 geeni mutatsiooniga a3-, a4-, a5- ja a6-ahelate puudumine IV tüüpi kollageeni struktuuris ning o1- ja a2-ahelate arv glomerulaarses basaalmembraanis suureneb. Selle nähtuse mehhanism on ebaselge, eeldatakse, et põhjuseks on mRNA posttranskriptsioonilised muutused.
Glomerulaarsete basaalmembraanide IV tüüpi kollageeni struktuuris a3, a4 ja a5 ahelate puudumine viib nende hõrenemiseni ja hapruseni Alporti sündroomi algstaadiumis, mis avaldub kliiniliselt sagedamini hematuuriana (harvemini hematuuriana koos proteinuuriaga või ainult proteinuuriaga), kuulmislanguse ja lentikoonusena. Haiguse edasine progresseerumine viib basaalmembraanide paksenemiseni ja läbilaskvuse halvenemiseni haiguse hilisemas staadiumis, kus neis vohab V ja VI tüüpi kollageen, mis avaldub proteinuuria suurenemises ja neerufunktsiooni languses.
Päriliku nefriidi aluseks oleva mutatsiooni olemus määrab suuresti selle fenotüübilise ilmingu. X-kromosoomi deletsiooni korral koos IV tüüpi kollageeni a5- ja a6-ahelate sünteesi eest vastutavate Col4A5 ja Col4A6 geenide samaaegse mutatsiooniga kaasneb Alporti sündroom söögitoru ja suguelundite leiomüomatoosiga. Uuringute andmetel täheldatakse deletsiooniga seotud Col4A5 geeni mutatsiooni korral patoloogilise protsessi suuremat raskusastet, neerukahjustuse kombinatsiooni ekstrarenaalsete ilmingutega ja kroonilise neerupuudulikkuse varajast arengut võrreldes selle geeni punktmutatsiooniga.
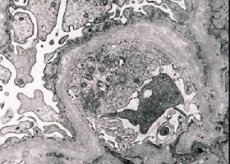
Morfoloogiliselt näitab elektronmikroskoopia glomerulaarsete basaalmembraanide (eriti lamina densa) hõrenemist ja kihistumist ning elektrontihedate graanulite olemasolu. Glomerulaarsed kahjustused võivad samal patsiendil olla heterogeensed, minimaalsetest fokaalsetest mesangiaalkahjustustest kuni glomeruloskleroosini. Alporti sündroomi korral on glomeruliit alati immunonegatiivne, mis eristab seda glomerulonefriidist. Iseloomulikeks tunnusteks on tubulaarse atroofia teke, lümfohistiotsütaarne infiltratsioon ja "vahtrakkude" olemasolu lipiidide kaasamisega - lipofaagidega. Haiguse progresseerumisel ilmneb glomerulaarsete basaalmembraanide paksenemine ja väljendunud hävimine.
Immuunsüsteemis ilmnevad teatud muutused. Päriliku nefriidiga patsientidel on IgA tase langenud ja kalduvus IgM kontsentratsiooni suurenemisele veres, IgG tase võib haiguse algstaadiumis olla suurenenud ja hilisemas staadiumis langeda. Võimalik, et IgM ja G kontsentratsiooni suurenemine on omamoodi kompenseeriv reaktsioon IgA puudulikkusele.
T-lümfotsüütide süsteemi funktsionaalne aktiivsus on vähenenud; täheldatakse Ig A sünteesi eest vastutavate B-lümfotsüütide selektiivset vähenemist, immuunsuse fagotsüütiline side on häiritud, peamiselt neutrofiilide kemotaksise ja rakusisese seedimisprotsesside häirete tõttu.
Alporti sündroomiga patsientidel neerubiopsia uurimisel näitavad elektronmikroskoopia andmed glomerulaarse basaalmembraani ultrastruktuurilisi muutusi: glomerulaarsete basaalmembraanide hõrenemine, struktuuri häire ja lõhenemine koos paksuse muutumise ja ebaühtlaste kontuuridega. Päriliku nefriidi algstaadiumis määrab defekt glomerulaarsete basaalmembraanide hõrenemise ja hapruse.
Glomeraarmembraanide hõrenemine on soodsam märk ja esineb sagedamini tüdrukutel. Päriliku nefriidi püsivam elektronmikroskoopiline tunnus on basaalmembraani lõhenemine ning selle hävimise raskusaste korreleerub protsessi raskusastmega.
Alporti sündroomi sümptomid lastel
Alporti sündroomi esimesed sümptomid isoleeritud kuseteede sündroomi kujul avastatakse kõige sagedamini esimese kolme eluaasta lastel. Enamasti avastatakse haigus juhuslikult. Kuseteede sündroom avastatakse lapse ennetava läbivaatuse käigus, enne lastehoiuasutusse vastuvõtmist või ARVI ajal. ARVI ajal uriinis patoloogia ilmnemisel. Päriliku nefriidi korral, erinevalt omandatud glomerulonefriidist, latentset perioodi ei ole.
Haiguse algstaadiumis lapse tervis ei kannata oluliselt, iseloomulikuks tunnuseks on kuseteede sündroomi püsivus ja vastupidavus. Üks peamisi tunnuseid on erineva raskusastmega hematuuria, mida täheldatakse 100% juhtudest. Hematuuria astme suurenemist täheldatakse hingamisteede infektsioonide, füüsilise koormuse või ennetavate vaktsineerimiste ajal või pärast neid. Proteinuuria ei ületa enamasti 1 g päevas, haiguse alguses võib see olla ebastabiilne, protsessi progresseerudes proteinuuria suureneb. Perioodiliselt võib uriinisettes esineda leukotsütuuriat lümfotsüütide ülekaaluga, mis on seotud interstitsiaalsete muutuste tekkega.
Seejärel halveneb neerufunktsioon osaliselt, patsiendi üldseisund halveneb: ilmneb joove, lihasnõrkus, arteriaalne hüpotensioon, sageli kuulmislangus (eriti poistel) ja mõnikord nägemislangus. Joove avaldub kahvatuse, väsimuse ja peavaludena. Haiguse algstaadiumis avastatakse kuulmislangus enamasti ainult audiograafia abil. Alporti sündroomi korral võib kuulmislangus esineda lapsepõlve erinevatel perioodidel, kuid kõige sagedamini diagnoositakse kuulmislangus 6-10-aastaselt. Laste kuulmislangus algab kõrgete sagedustega, saavutades olulise õhu- ja luujuhtivuse, liikudes heli juhtivast kuulmislangusest heli tajuva kuulmislanguseni. Kuulmislangus võib olla üks haiguse esimesi sümptomeid ja võib eelneda kuseteede sündroomile.
20% juhtudest esineb Alporti sündroomiga patsientidel muutusi nägemisorganites. Kõige sagedamini tuvastatakse läätse anomaaliaid: sferofookiat, eesmist, tagumist või segatüüpi läätsekoonust ja mitmesugused kataraktid. Alporti sündroomiga perekondades esineb lühinägelikkust märkimisväärselt. Mitmed teadlased märgivad nendes perekondades pidevalt kahepoolseid perimakulaarseid muutusi erkvalkjate või kollakate granulatsioonide kujul kollaskehas. Nad peavad seda tunnust püsivaks sümptomiks, millel on Alporti sündroomi korral kõrge diagnostiline väärtus. K. S. Chugh jt (1993) leidsid oftalmoloogilises uuringus Alporti sündroomiga patsientidel nägemisteravuse langust 66,7% juhtudest, eesmist läätsekoonust 37,8% juhtudest, võrkkesta laike 22,2% juhtudest, katarakti 20% juhtudest ja keratokonust 6,7% juhtudest.
Mõnedel päriliku nefriidiga lastel, eriti neerupuudulikkuse tekkimisel, on täheldatud märkimisväärset füüsilise arengu mahajäämust. Neerupuudulikkuse progresseerumisel tekib arteriaalne hüpertensioon. Lastel avastatakse seda sagedamini noorukieas ja vanemates vanuserühmades.
Päriliku nefriidiga patsientidele on iseloomulik mitmesuguste (rohkem kui 5-7) sidekoe düsmorfogeneesi stigmade esinemine. Patsientide sidekoe stigmade hulgas on kõige levinumad silmade hüpertelorism, kõrge suulagi, hammustuse anomaaliad, kõrvakeste ebanormaalne kuju, väikese sõrme kõverus kätel ja "sandaalivahe" jalgadel. Pärilikku nefriiti iseloomustab düsmorfogeneesi stigmade ühtlus perekonnas, samuti nende suur jaotumine sugulaste vahel, kelle liini mööda haigus levib.
Haiguse algstaadiumis tuvastatakse neerude osaliste funktsioonide isoleeritud langus: aminohapete transport, elektrolüüdid, kontsentratsioonifunktsioon, atsidogenees, hilisemad muutused mõjutavad nii nefroni proksimaalse kui ka distaalse osa funktsionaalset seisundit ja neid iseloomustavad kombineeritud osalised häired. Glomerulaarfiltratsiooni langus tekib hiljem, sagedamini noorukieas. Päriliku nefriidi progresseerumisel tekib aneemia.
Seega iseloomustab pärilikku nefriiti haiguse järkjärguline kulg: esiteks latentne staadium ehk varjatud kliinilised sümptomid, mis avalduvad kuseteede sündroomi minimaalsete muutustena, seejärel toimub protsessi järkjärguline dekompensatsioon neerufunktsiooni langusega koos manifestsete kliiniliste sümptomitega (mürgistus, asteenia, arengupeetus, aneemia). Kliinilised sümptomid ilmnevad tavaliselt olenemata põletikulise reaktsiooni kihistumisest.
Pärilik nefriit võib avalduda erinevatel vanuseperioodidel, mis sõltub geeni toimest, mis on teatud ajani represseeritud olekus.
Klassifikatsioon
Pärilikku nefriiti on kolme tüüpi
- I variant - kliiniliselt avaldub nefriidina koos hematuuria, kuulmislanguse ja silmakahjustusega. Nefriidi kulg on progresseeruv kroonilise neerupuudulikkuse tekkega. Pärilikkuse tüüp on dominantne, seotud X-kromosoomiga. Morfoloogiliselt ilmneb basaalmembraani struktuuri häire, selle hõrenemine ja lõhenemine.
- II variant - kliiniliselt avaldub nefriidina koos hematuuriaga ilma kuulmislanguseta. Nefriidi kulg on progresseeruv kroonilise neerupuudulikkuse tekkega. Pärilikkuse tüüp on dominantne, seotud X-kromosoomiga. Morfoloogiliselt tuvastatakse glomerulaarsete kapillaaride basaalmembraani (eriti laminadensa) hõrenemine.
- III variant - healoomuline perekondlik hematuuria. Kulg on soodne, kroonilist neerupuudulikkust ei teki. Pärilikkuse tüüp on autosomaalselt dominantne või autosomaalselt retsessiivne. Autosomaalselt retsessiivse päranditüübi korral on naistel haiguse raskem kulg.
Alporti sündroomi diagnoosimine
Pakutakse välja järgmised kriteeriumid:
- vähemalt kahe nefropaatiaga patsiendi olemasolu igas perekonnas;
- hematuuria kui probandi nefropaatia peamine sümptom;
- kuulmislanguse esinemine vähemalt ühel pereliikmel;
- kroonilise neerupuudulikkuse teke ühel või mitmel sugulasel.
Erinevate pärilike ja kaasasündinud haiguste diagnoosimisel pööratakse suurt tähelepanu terviklikule lähenemisele uurimisele ja ennekõike lapse sugupuu koostamisel saadud andmetele. Alporti sündroomi diagnoosi peetakse kehtivaks juhtudel, kui patsiendil tuvastatakse 3 neljast tüüpilisest tunnusest: hematuuria ja kroonilise neerupuudulikkuse esinemine perekonnas, neurosensorse kuulmislanguse esinemine, nägemise patoloogia patsiendil, glomerulaarse basaalmembraani lõhustumise tunnuste tuvastamine koos selle paksuse muutusega ja ebaühtlaste kontuuridega biopsia elektronmikroskoopiliste omaduste hindamisel.
Patsiendi läbivaatus peaks hõlmama kliinilisi ja geneetilisi uurimismeetodeid; haiguse anamneesi sihipärast uurimist; patsiendi üldist läbivaatust, võttes arvesse diagnostiliselt olulisi kriteeriume. Kompensatsioonistaadiumis saab patoloogiat tuvastada ainult selliste sündroomide uurimisega nagu päriliku koormuse olemasolu, hüpotensioon, düsembrüogeneesi mitmed stigmad, kuseteede sündroomi muutused. Dekompensatsioonistaadiumis võivad ilmneda neerudevälised sümptomid, nagu raske joove, asteenia, füüsilise arengu aeglustumine, aneemia, mis avalduvad ja süvenevad neerufunktsiooni järkjärgulise langusega. Enamikul patsientidest täheldatakse neerufunktsiooni langusega järgmist: atsido- ja aminogeneesi vähenemine; 50% patsientidest märgib neerude sekretoorse funktsiooni olulist langust; uriini optilise tiheduse kõikumiste piiratud ulatust; filtratsioonirütmi häireid ja seejärel glomerulaarfiltratsiooni vähenemist. Kroonilise neerupuudulikkuse staadium diagnoositakse, kui patsientidel on vereseerumis kõrgenenud uurea tase (üle 0,35 g/l) 3-6 kuud või kauem ja glomerulaarfiltratsioon on vähenenud 25%-ni normist.
Päriliku nefriidi diferentsiaaldiagnostikat tuleks teostada eelkõige omandatud glomerulonefriidi hematuurse vormi korral. Omandatud glomerulonefriidil on kõige sagedamini äge algus, 2-3 nädalat pärast infektsiooni, neerupealiste tunnused, sealhulgas hüpertensioon esimestest päevadest alates (päriliku nefriidi korral vastupidi hüpotensioon), glomerulaarfiltratsiooni vähenemine haiguse alguses, osaliste tubulaarsete funktsioonide häireid ei esine, samas kui päriliku nefriidi korral need esinevad. Omandatud glomerulonefriit esineb väljendunuma hematuuria ja proteinuuriaga ning suurenenud erütrotsüütide settimiskiirusega (ESR). Diagnostilise väärtusega on tüüpilised muutused glomerulaarse basaalmembraanis, mis on iseloomulikud pärilikule nefriidile.
Düsmetaboolse nefropaatia diferentsiaaldiagnostikat tehakse kroonilise neerupuudulikkuse korral, perekonnas on kliiniliselt tuvastatud heterogeenseid neeruhaigusi ning nefropaatia spekter võib olla püelonefriidist urolitiaasini. Lastel esineb sageli kaebusi kõhuvalu kohta ja perioodiliselt urineerimisel, uriinis on oksalaate.
Päriliku nefriidi kahtluse korral tuleb patsient diagnoosi selgitamiseks suunata spetsialiseeritud nefroloogiaosakonda.
Mida tuleb uurida?
Millised testid on vajalikud?
Kellega ühendust võtta?
Alporti sündroomi ravi
Režiim hõlmab piiranguid raskele füüsilisele koormusele ja värske õhu viibimisele. Toitumine on täisväärtuslik, piisava koguse täisväärtuslike valkude, rasvade ja süsivesikutega, arvestades neerufunktsiooni. Suur tähtsus on krooniliste infektsioonikollete avastamisel ja ravimisel. Kasutatakse järgmisi ravimeid: ATP, kokarboksülaas, püridoksiin (kuni 50 mg/päevas), karnitiinkloriid. Kuure manustatakse 2-3 korda aastas. Hematuuria korral on ette nähtud taimne ravim - nõges, arooniamahl, raudrohi.
Välis- ja kodumaises kirjanduses on teateid prednisolooniga ravi ja tsütostaatikumide kasutamise kohta. Selle mõju on aga raske hinnata.
Kroonilise neerupuudulikkuse korral kasutatakse hemodialüüsi ja neerusiirdamist.
Päriliku nefriidi spetsiifilise (tõhusa patogeneetilise) ravi meetodid puuduvad. Kõik ravimeetmed on suunatud neerufunktsiooni languse ennetamisele ja aeglustamisele.
Toitumine peaks olema tasakaalustatud ja kõrge kalorsusega, võttes arvesse neerude funktsionaalset seisundit. Funktsionaalsete häirete puudumisel peaks lapse toit sisaldama piisavalt valke, rasvu ja süsivesikuid. Neerufunktsiooni häire tunnuste esinemisel tuleks piirata valkude, süsivesikute, kaltsiumi ja fosfori hulka, mis lükkab edasi kroonilise neerupuudulikkuse teket.
Füüsiline aktiivsus peaks olema piiratud; lastel soovitatakse sportimist vältida.
Vältida tuleks kokkupuudet nakkushaigustega patsientidega, vähendada ägedate hingamisteede haiguste tekkeriski. Vajalik on kroonilise infektsiooni fookuste puhastamine. Päriliku nefriidiga lastele ennetavaid vaktsineerimisi ei tehta, vaktsineerimine on võimalik ainult epidemioloogiliste näidustuste korral.
Päriliku nefriidi korral on hormonaalne ja immunosupressiivne ravi ebaefektiivne. Tsüklosporiin A ja AKE inhibiitorite pikaajalisel mitmeaastasel kasutamisel on märke teatud positiivsest mõjust (proteinuuria vähenemine ja haiguse progresseerumise aeglustumine).
Patsientide ravis kasutatakse ainevahetust parandavaid ravimeid:
- püridoksiin - 2-3 mg/kg/päevas 3 annusena 4 nädala jooksul;
- kokarboksülaas - 50 mg intramuskulaarselt ülepäeviti, kokku 10-15 süsti;
- ATP - 1 ml intramuskulaarselt ülepäeviti, 10-15 süsti;
- A-vitamiin - 1000 RÜ/aastas/päevas ühe annusena 2 nädala jooksul;
- E-vitamiin - 1 mg/kg/päevas ühe annusena 2 nädala jooksul.
Seda tüüpi ravi aitab parandada patsientide üldist seisundit, vähendada tubulaarseid düsfunktsioone ja seda viiakse läbi kursustel 3 korda aastas.
Levamisooli saab kasutada immunomodulaatorina - 2 mg/kg/päevas 2-3 korda nädalas, annuste vahel 3-4 päeva.
Uuringute andmete kohaselt on hüperbaarilisel hapnikuga varustamisel positiivne mõju hematuuria ja neerufunktsiooni häirete raskusastmele.
Päriliku nefriidi ravi kõige efektiivsem meetod on õigeaegne neerusiirdamine. Sellisel juhul ei esine siirdamisel haiguse retsidiivi; väikesel protsendil juhtudest (umbes 5%) võib siirdatud neerus tekkida nefriit, mis on seotud glomerulaarse basaalmembraani antigeenidega.
Paljutõotav suund on sünnieelne diagnostika ja geenitehnoloogia. Loomkatsed näitavad IV tüüpi kollageeni alfa-ahelate sünteesi eest vastutavate normaalsete geenide neerukoesse ülekandmise suurt efektiivsust, mille järel täheldatakse normaalsete kollageenistruktuuride sünteesi.
Prognoos
Päriliku nefriidi prognoos on alati tõsine.
Päriliku nefriidi kulgu prognostiliselt ebasoodsad kriteeriumid on:
- meessugu;
- kroonilise neerupuudulikkuse varajane areng pereliikmetel;
- proteinuuria (rohkem kui 1 g päevas);
- glomerulaarsete basaalmembraanide paksenemine mikroskoopia abil;
- akustiline neuriit;
- deletsioon Col4A5 geenis.
Healoomulise perekondliku hematuuria prognoos on soodsam.
Использованная литература