Uued väljaanded
Trigonookaalia
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
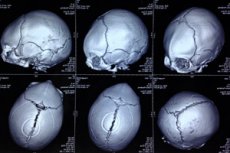
Kolju deformatsiooni vormis esinevat kaasasündinud anomaaliat, mille puhul imikute pea on ebakorrapärase kujuga ja kolju näib kolmnurkne, defineeritakse kui trigonokefaaliat (kreeka keelest trigonon – kolmnurk ja kephale – pea). [ 1 ]
Epidemioloogia
Kraniosünostoosi esinemissagedus on hinnanguliselt ligikaudu viis juhtu 10 000 elussünni kohta (või üks juhtum 2000–2500 inimese kohta üldpopulatsioonis). [ 2 ]
85% juhtudest on kraniosünostoos juhuslik, ülejäänud juhud esinevad osana sündroomist. [ 3 ]
Statistika kohaselt on keskmise otsmikuõmbluse enneaegne sulandumine kraniosünostoosi esinemissageduselt teine vorm ning trigonokefaaliat esineb ühel juhul 5–15 tuhande vastsündinu kohta; selle anomaaliaga poisslapsi on peaaegu kolm korda rohkem kui tüdrukuid. [ 4 ]
Ligikaudu 5% juhtudest esineb see kaasasündinud anomaalia perekonnaanamneesis. [ 5 ]
Põhjused trigonookaalia
Kolju normaalne moodustumine toimub tänu selles paiknevatele primaarsetele kasvu- ja luude ümberehituskeskustele – kraniofakiaalsetele sünartroosidele (liigeste) –, mis sulguvad pea skeleti arengu ajal teatud ajal, tagades luude kokkukasvamise. [ 6 ]
Vastsündinu kolju otsmikuluu (os frontale) koosneb kahest poolest, mille vahel on vertikaalne kiuline ühendus – mediaanne otsmiku- ehk metoopiline õmblus (kreeka keelest metopon – laup), mis kulgeb ninaselja ülaosast mööda lauba keskjoont ülespoole eesmise fontanellini. See on ainus kiuline koljuõmblus, mis paraneb imikueas: 3–4 kuust kuni 8–18-aastaseni. [ 7 ]
Vaata ka – Kolju muutused pärast sündi
Trigonokefaalia põhjused on metoopiline kraniosünostoos (kraniosünostoos) või metoopiline sünostoos (kreeka keelest syn – koos ja osteon – luu), see tähendab koljuvõlvi luude enneaegset (enne kolmandat kuud) liikumatut kokkukasvamist piki keskmist otsmikuõmblust. Seega on kraniosünostoos ja trigonokefaalia seotud põhjuse ja tagajärje või patoloogilise protsessi ja selle tagajärjena. [ 8 ]
Enamasti on lapse trigonotsefaalia primaarse (isoleeritud) kraniosünostoosi tagajärg, mille täpne põhjus on teadmata. Isoleeritud kraniosünostoosi esineb juhuslikult, tõenäoliselt geneetiliste ja keskkonnategurite kombinatsiooni tõttu. [ 9 ]
Kuid trigonokefaalia võib olla osa kaasasündinud sündroomidest, mis tekivad kromosomaalsete kõrvalekallete ja erinevate geenide mutatsioonide tõttu. Nende hulka kuuluvad: Opitzi trigonokefaalia sündroom (Bohring-Opitzi sündroom), Aperti sündroom, Loeys-Dietzi sündroom, Pfeifferi sündroom, Jackson-Weissi sündroom, kraniofakiaalne düsostoos või Crouzoni sündroom, Jacobseni, Saethre-Chotzeni, Münke sündroomid. Sellistel juhtudel nimetatakse trigonokefaaliat sündroomseks. [ 10 ]
Sündides on aju suurus tavaliselt 25% täiskasvanu suurusest ja esimese eluaasta lõpuks ulatub see umbes 75%-ni täiskasvanu aju suurusest. Kuid aju kasvu primaarse hilinemise korral on võimalik nn sekundaarne kraniosünostoos. Hilinemise etioloogia on seotud ainevahetushäirete, mõnede hematoloogiliste haiguste ja kemikaalide (sealhulgas ravimites sisalduvate) teratogeense toimega lootele. [ 11 ]
Ekspertide sõnul püsib täiskasvanutel, keda lapsepõlves ei ravitud isoleeritud kraniosünostoosi või kaasasündinud sündroomi tagajärjel, trigonotsefaalia kogu elu. [ 12 ]
Riskitegurid
Eksperdid usuvad, et trigonokefaalia (ja selle põhjusena metoopilise kraniosünostoosi) peamised riskifaktorid on geneetilised: viimase kahe aastakümne jooksul on tuvastatud üle 60 geeni, mille mutatsioonid on seotud kolju luude enneaegse ja liikumatu kokkukasvamisega imikutel.
Kraniofakiaalse sünartroosi ja üldise osteogeneesi (luukoe moodustumise) patoloogiate tekkerisk suureneb loote ebanormaalse esituse, emakasisese hüpoksia, mitmikraseduse, alkoholitarbimise, narkootikumide tarvitamise või suitsetamise korral raseduse ajal. [ 13 ]
Pathogenesis
Valitseva teooria kohaselt on trigonokefaalia patogeneesi aluseks loote osteogeneesi häire raseduse algstaadiumis, mille põhjuseks on enamasti geneetilised tegurid, kuna metoopilise kraniosünostoosiga vastsündinutel avastatakse juhuslikke kromosomaalseid kõrvalekaldeid. Näiteks üks levinumaid on trisoomia 9p, mis põhjustab kraniofakiaalseid ja skeleti defekte, vaimset alaarengut ja psühhomotoorset arenguhäireid. [ 14 ]
Keskmise otsmikuõmbluse liiga varajase kokkukasvamise tõttu on selle koljuosa kasv takistatud: otsmikuluu külgmine kasv on piiratud eesmise koljulohu lühenemise tõttu; otsaesise keskjoonele moodustub luuline harja; toimub silmakoopaid moodustavate luude koondumine ja oimusluude süvend. [ 15 ]
Kuid kolju kasv teistes piirkondades jätkub: toimub kolju tagaosa kompenseeriv sagitaalne (ees-taga) ja põikisuunaline kasv (koos parietaal-kuklaluu osa laienemisega), samuti näo ülaosa vertikaalne ja sagitaalne kasv. Nende häirete tagajärjel omandab kolju ebakorrapärase kuju - kolmnurkse.
Sümptomid trigonookaalia
Trigonokefaalia peamised sümptomid on pea kuju ja välimuse muutused:
- pea kroonist ülalt vaadatuna on kolju kolmnurga kujuline;
- kitsenenud otsmik;
- märgatav või palpeeritav seljandik (luine eend), mis kulgeb mööda otsaesise keskosa ja annab otsmikuluule terava (kiilukujulise) välimuse;
- silmakoopa ülemise osa deformatsioon (silmakoopa ülaosa lamenemine) ja hüpotelorism (silmadevahelise kauguse vähenemine).
Samuti võib eesmine (eesmine) fontanelle enneaegselt sulguda.
Sündroomse trigonokefaalia korral esineb lastel ka teisi anomaaliaid ja vaimse alaarengu tunnuseid. [ 16 ]
Diagnostika trigonookaalia
Trigonokefaaliat diagnoositakse sünnihetkel või mõne kuu jooksul pärast sündi. Metoopilise kraniosünostoosi vähem rasked tunnused ei pruugi aga ilmneda enne varast lapsepõlve.
Kolju patoloogia visualiseerimiseks viiakse läbi instrumentaalne diagnostika, kasutades pea kompuutertomograafiat ja ultraheli. [ 19 ], [ 20 ]
Diferentseeritud diagnoos
Sündroomse defekti eristamiseks isoleeritud metopilise sünostoosist on vajalik diferentsiaaldiagnostika, mille puhul lapsele tehakse genotüübi test.
Ravi trigonookaalia
Mõnedel lastel on metoopilise sünostoosi juhud üsna kerged (kui otsmikul on ainult märgatav vagu ja muid sümptomeid pole) ega vaja spetsiifilist ravi. [ 21 ]
Raske trigonokefaalia ravi on kirurgiline ja hõlmab operatsiooni pea kuju korrigeerimiseks ja normaalse aju kasvu tagamiseks, samuti näoluude deformatsioonide kirurgilist korrigeerimist. [ 22 ]
See kirurgiline protseduur – metoopilise õmbluse sünosteektoomia, silmakoopa ääre nihutamine ja kranioplastika – tehakse enne 6 kuu vanuseks saamist. Last jälgitakse kuni üheaastaseks saamiseni; esimestel eluaastatel uuritakse last perioodiliselt, et veenduda kõne, motoorsete oskuste või käitumise probleemide puudumises. [ 23 ]
Ärahoidmine
Selle kaasasündinud defekti ennetamiseks pole meetodeid, kuid geneetiline nõustamine võib ära hoida ravimatu kraniotserebraalse patoloogiaga lapse sündi.
Loote kraniosünostoosi saab tuvastada raseduse teisel ja kolmandal trimestril pea sünnieelse ultraheli abil.
Prognoos
Prognoos sõltub suuresti kolju deformatsiooni astmest, mis mõjutab aju neurokognitiivseid funktsioone. Ja kui korrektiivset operatsiooni pole tehtud, on trigonotsefaaliaga lastel – võrreldes tervete eakaaslastega – madalamad üldised kognitiivsed võimed, probleemid kõne, nägemise, tähelepanu ja käitumisega.
Использованная литература