Artikli meditsiiniline ekspert

Uued väljaanded
Achondroplaasia
Last reviewed: 12.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
On palju haruldasi kaasasündinud haigusi ja üks neist on luu kasvu rikkumine - akondroplaasia, mis põhjustab tõsist ebaproportsionaalset lühikest kasvu.
RHK-10 arenguanomaaliate osas on seda tüüpi päriliku osteokondraalse düsplaasia koos torukujuliste luude ja selgroo kasvudefektidega kood Q77.4 [ 1 ].
Epidemioloogia
Akondroplaasia levimuse osas on erinevate uuringute statistilised andmed mitmetähenduslikud. Mõned väidavad, et see anomaalia esineb ühel vastsündinul 10 000-st, teised - ühel 26-28 000-st ja veel teised - 4-15 juhul 100 000-st. [ 2 ]
Samuti on teavet, et kui isa on üle 50 aasta vana, on akondroplaasia esinemissagedus lastel üks juhtum 1875 vastsündinu kohta.
Põhjused achondroplaasia
Akondroplaasia põhjuseks on osteogeneesi rikkumine, eriti üks skeleti torukujuliste luude diafüüsi emakasisese luustumise tüüpidest - endokondraalne luustumine, mille käigus kõhre modifitseeritakse luukoeks. Lisateavet leiate jaotisest - Luude areng ja kasv.
Pikkade luude luustumise häire ehk loote akondroplaasia tekib membraantürosiinkinaasi geeni - fibroblastide kasvufaktori retseptori 3 (FGFR3 kromosoomis 4p16.3) mutatsioonide tõttu, mis mõjutavad rakkude kasvu ja diferentseerumist. FGFR3 mutatsioonide esinemine on seotud geneetilise ebastabiilsuse ja kromosoomide arvu muutustega (aneuploidia).
Akondroplaasia kandub lapsele edasi autosomaalselt dominantselt, see tähendab, et ta saab ühe koopia mutantsest geenist (mis on dominantne) ja ühe normaalse geeni paaril mittesoolisel (autosomaalsel) kromosoomil. Seega on selle defekti pärandumisviis autosomaalselt dominantne ja anomaalia võib avalduda 50% järglastest, kui selle geeni (genotüübi) alleelide kombinatsioon ristub.
Lisaks võivad mutatsioonid olla juhuslikud ja nagu praktika näitab, sünnib 80% juhtudest akondroplaasiaga lapsed normaalse pikkusega vanematele.
Riskitegurid
Akondroplaasiaga laste sünni peamised riskifaktorid on pärilikud. Kui ühel vanematest on see defekt, siis haige lapse saamise tõenäosus on hinnanguliselt 50%; kui mõlemal vanemal on see anomaalia, on see samuti 50%, kuid homosügootse akondroplaasia risk on 25%, mis viib surmani enne sündi või varases imikueas.
Isa vanuse kasvades (lähemal 40. eluaastal ja vanemal) suureneb FGFR3 geeni uue mutatsiooni (de novo mutatsiooni) risk.
Pathogenesis
Akondroplaasia patogeneesi selgitades rõhutavad eksperdid transmembraanse valgu türosiinproteiinkinaasi (kodeerib FGFR3 geen) olulisust kasvuplaatide kõhrekoe rakkude - kondrotsüütide - jagunemise, diferentseerumise ja apoptoosi reguleerimisel, samuti skeleti normaalse arengu - osteogeneesi ja luukoe mineralisatsiooni - tagamisel.
Embrüonaalse arengu käigus geenimutatsiooni korral muutuvad fibroblastkasvufaktori 3 retseptorid aktiivsemaks. Nende funktsioonide suurenemine häirib rakuliste signaalide ülekannet ja selle valgu rakuvälise osa interaktsiooni polüpeptiidsete fibroblastkasvufaktoritega (FGF). Selle tulemusena tekib ebaõnnestumine: kõhrerakkude proliferatsiooni etapp lüheneb ja nende diferentseerumine algab oodatust varem. Kõik see viib kolju luude ebaõige moodustumise ja kokkukasvamiseni ning skeleti düsplaasiani - pikkade luude vähenemiseni, millega kaasneb väljendunud lühike kasv ehk kääbuskasvulisus.
Ja kaks kolmandikku kääbuskasvu juhtudest on seotud akondroplaasiaga.
Sümptomid achondroplaasia
Ebanormaalne luukasv põhjustab akondroplaasia kliinilisi sümptomeid, näiteks:
- väljendunud lühike kasv (ebaproportsionaalne kääbuskasvulisus) keskmise täiskasvanu pikkusega 123–134 cm;
- ala- ja ülajäsemete proksimaalsete osade lühenemine suhteliselt normaalse torso suurusega;
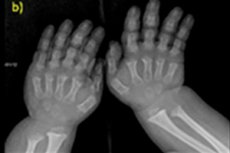
- lühenenud sõrmed ja varbad;
- suurenenud pea (makro- või megalotsefaalia); [ 3 ]
- spetsiifilised näojooned väljaulatuva otsaesise ja näo keskosa hüpoplaasia kujul - nina sisse vajunud sild.
- kitsas kraniokaela ühenduskoht. Mõned akondroplaasiaga imikud surevad esimesel eluaastal kraniokaela ühenduskohaga seotud tüsistuste tõttu; populatsiooniuuringud näitavad, et see surmaoht võib ilma hindamise ja sekkumiseta ulatuda kuni 7,5%-ni.[ 4 ]
- Keskkõrva düsfunktsioon on sageli probleem [ 5 ] ja kui seda ei ravita korralikult, võib see viia juhtiva kuulmislanguse tekkeni, mis on piisavalt tõsine, et häirida kõne arengut. Enam kui pooled lastest vajavad rõhuühtlustustoru. [ 6 ] Üldiselt on umbes 40%-l akondroplaasiaga inimestest funktsionaalselt oluline kuulmislangus. Ka ekspressiivse keele areng on sageli hilinenud, kuigi kuulmislanguse ja ekspressiivse keele probleemide vahelise seose tugevus on küsitav.
- Sääreluu kõverdumine on akondroplaasiaga inimestel väga levinud. Üle 90%-l ravimata täiskasvanutest esineb mingil määral kõverdumist.[ 7 ] „Kõverdumine” on tegelikult keeruline deformatsioon, mis tuleneb külgkallutuse, sääreluu sisemise torsiooni ja põlve dünaamilise ebastabiilsuse kombinatsioonist.[ 8 ]
Akondroplaasiaga imikutele on iseloomulik lihashüpotoonia, mille tõttu hakkavad nad liikumisoskusi ja kõndimist omandama hiljem. Intellekti ja kognitiivseid võimeid see arenguhäire ei mõjuta. [ 9 ], [ 10 ]
Tagajärjed ja tüsistused
Seda tüüpi pärilikku osteokondraalset düsplaasiat iseloomustavad järgmised tüsistused ja tagajärjed:
- korduvad kõrvapõletikud;
- obstruktiivne uneapnoe;
- hüdrotsefaalia;
- hambumushäired ja kõverad hambad:
- jalgade deformatsioon (varus või valgus) koos kõnnaku muutusega;
- nimmelülide hüpertroofiline lordoos või selle kõverus (rinda-nimmepiirkonna küfoos või nimmepiirkonna skolioos) - seljavaluga kõndimisel;
- liigesevalu (luude vale asendi või närvijuurte kokkusurumise tõttu);
- Seljaaju stenoos ja seljaaju kokkusurumine; Täiskasvanueas on kõige levinum meditsiiniline kaebus sümptomaatiline seljaaju stenoos, mis hõlmab L1-L4. Sümptomid ulatuvad vahelduvast, pöörduvast lonkamisest, mis on põhjustatud treeningust, kuni raskete, pöördumatute jalgade talitlushäirete ja uriinipeetuseni.[ 11 ] Lonkamine ja stenoos võivad põhjustada nii sensoorseid (tuimus, valu, raskustunne) kui ka motoorseid sümptomeid (nõrkus, komistamine, piiratud kõndimistaluvus). Vaskulaarne lonkamine tuleneb veresoonte tursest pärast seismist ja kõndimist ning on puhkuse ajal täielikult pöörduv. Seljaaju stenoos on seljaaju või närvijuure tegelik kahjustus seljaajukanali stenootilise luu poolt ja sümptomid on pöördumatud. Konkreetsele dermatoomile lokaliseeritud sümptomid võivad tuleneda spetsiifiliste närvijuure avade stenoosist.
- rindkere seina vähenemine koos piiratud kopsukasvu ja kopsufunktsiooni langusega (raske õhupuudus). Imikueas on väikesel rühmal akondroplaasiaga inimestel restriktiivsed kopsuprobleemid. Väikesed rinnad ja suurenenud rindkere elastsus koos põhjustavad kopsumahu vähenemist ja restriktiivset kopsuhaigust [ 12 ].
Muud ortopeedilised probleemid
- Liigeste nõrkus. Enamik liigeseid on lapsepõlves hüpermobiilsed. Üldiselt on sellel vähe mõju, välja arvatud põlve ebastabiilsus mõnedel inimestel.
- Diskoidne lateraalne menisk: see hiljuti tuvastatud struktuuriline anomaalia võib mõnedel inimestel põhjustada kroonilist põlvevalu.[ 13 ]
- Artriit: FGFR-3 konstitutiivne aktivatsioon, nagu akondroplaasia korral, võib kaitsta artriidi tekke eest.[ 14 ]
- Acanthosis nigricans'i esineb ligikaudu 10%-l akondroplaasiaga inimestest.[ 15 ] Selles populatsioonis ei peegelda see hüperinsulinemiat ega pahaloomulisust.
FGFR3 geeni 1138. nukleotiidi bialleelsete patogeensete variantide põhjustatud homosügootne akondroplaasia on raske haigus, mille radioloogilised leiud erinevad kvalitatiivselt akondroplaasia korral täheldatutest. Varajane surm tuleneb hingamispuudulikkusest, mis on tingitud väikesest rindkere seinast, ja neuroloogilistest defitsiitidest, mis on tingitud emakakaela-medullaarsest stenoosist [Hall 1988].
Diagnostika achondroplaasia
Enamikul patsientidest pannakse akondroplaasia diagnoos iseloomulike kliiniliste tunnuste ja radiograafiliste leidude põhjal. Imikutel või mõnede sümptomite puudumisel kasutatakse lõpliku diagnoosi panemiseks geneetilist testimist, näiteks karüotüübi analüüsi.[ 16 ]
Molekulaargeneetilise meetodi abil sünnieelse diagnostika tegemisel saab teha lootevee või koorionivilla proovi analüüse.
Loote ultraheliuuringul esinevad akondroplaasia tunnused - jäsemete lühenemine ja tüüpilised näojooned - visualiseeritakse pärast 22. rasedusnädalat.
Instrumentaalne diagnostika hõlmab ka skeleti röntgenülesvõtet või luude ultraheli. Röntgen kinnitab diagnoosi selliste andmete põhjal nagu suur kolju kitsa kuklaluu ava ja suhteliselt väikese alusega; lühikesed torukujulised luud ja lühenenud ribid; lühikesed ja lamedad lülikehad; ahenenud selgrookanal, niudeluu tiibade väiksem suurus.
Diferentseeritud diagnoos
Hüpofüüsi kääbuskasvu, kaasasündinud spondüloepifüüsi ja diastroofse düsplaasia, hüpokondroplaasia, Šereshevski-Turneri ja Noonani sündroomide ning pseudoakondroplaasia diferentsiaaldiagnostika on vajalik. Seega seisneb pseudoakondroplaasia ja akondroplaasia erinevus selles, et pseudoakondroplaasia korral on kääbuskasvuga patsientidel pea suurus ja näojooned normaalsed.
Kellega ühendust võtta?
Ravi achondroplaasia
Ameerika Pediaatria Akadeemia Geneetikakomitee on välja töötanud akondroplaasiaga laste ravi soovitused. Need soovitused on mõeldud suuniste andmiseks ega asenda individuaalset otsustusprotsessi. Hiljutine ülevaade [Pauli & Botto 2020] sisaldab ka juhiseid. On olemas erialakliinikud, mis on spetsialiseerunud skeleti düsplaasia ravile; nende soovitused võivad neist üldistest soovitustest veidi erineda.
Soovitused hõlmavad (kuid mitte ainult) järgmist.
Hüdrotsefaalia. Kui ilmnevad koljusisese rõhu tõusu tunnused või sümptomid (nt kiirenenud pea kasv, püsivalt punnis fontanell, näo pindmiste veenide märgatav suurenemine, ärrituvus, oksendamine, nägemishäired, peavalu), on vajalik suunata patsient neurokirurgi juurde.
Akondroplaasia korral esineva hüdrotsefaali oletatav etioloogia on suurenenud koljusisene venoosne rõhk, mis on tingitud kaelaava stenoosist. Seetõttu on standardseks raviks olnud ventrikuloperitoneaalne šunteerimine. Siiski võib endoskoopiline kolmas ventrikulostoomia mõnedel inimestel olla kasulik,[ 17 ] mis viitab sellele, et võivad olla kaasatud ka muud mehhanismid, näiteks neljanda vatsakese väljavoolu takistus kraniokervikaalse stenoosi tõttu.[ 18 ]
Kraniocervikaalse ülemineku stenoos. Suboktsipitaalse dekompressiooni vajaduse parimad ennustajad:
- Alajäsemete hüperrefleksia või kloonus
- Keskne hüpopnea polüsomnograafial
- Kolju- ja kaelapiirkonna kompuutertomograafia abil määratud foramen magnumi suuruse vähenemine ja võrdlus akondroplaasiaga laste normidega.[ 19 ]
- Hiljuti on operatsiooni otsustamisel arvestatava tegurina pakutud välja seljaaju kokkusurumise ja/või T2-kaalutud signaali kõrvalekallete tõendeid.
Kui esinevad selged sümptomaatilise kompressiooni tunnused, tuleb patsient dekompressioonikirurgiasse kiiresti suunata laste neurokirurgi juurde. [ 20 ]
Obstruktiivse uneapnoe ravi võib hõlmata järgmist:
- Adenotonsillektoomia
- Positiivne hingamisteede rõhk
- Trahheostoomia äärmuslikel juhtudel
- Kaalulangus
Need sekkumised võivad leevendada unehäireid ja parandada neuroloogilist funktsiooni.[ 21 ]
Harvadel juhtudel, kui obstruktsioon on piisavalt tõsine, et vajada trahheostoomiat, on ülemiste hingamisteede obstruktsiooni leevendamiseks kasutatud näo keskosa ettepoole suunatud operatsiooni.[ 22 ]
Keskkõrva düsfunktsioon. Sagedasi keskkõrvapõletikke, püsivat keskkõrvavedelikku ja sellest tulenevat kuulmislangust tuleks vajadusel agressiivselt ravida. Soovitatav on pikaajaline toru kasutamine, kuna seda on sageli vaja kuni seitsme- või kaheksa-aastaseks saamiseni.[ 23 ]
Kui probleemid tekivad igas vanuses, on soovitatav kasutada sobivaid ravimeetodeid.
Lühike kasv. Mitmed uuringud on hinnanud kasvuhormooni (GH) ravi kui võimalikku ravi lühikese kasvu akondroplaasia korral.[ 24 ]
Üldiselt näitavad need ja teised seeriad esialgset kasvu kiirenemist, kuid selle mõju väheneb aja jooksul.
Keskmiselt võib täiskasvanuna oodata vaid umbes 3 cm pikkuse kasvu.
Jäsemete pikendamine erinevate tehnikate abil on mõne jaoks endiselt võimalik. Võimalik on saavutada kuni 30–35 cm pikkuse kasvu. [ 25 ] Tüsistused on tavalised ja võivad olla tõsised.
Kuigi mõned pooldavad nende protseduuride tegemist juba kuue kuni kaheksa-aastaselt, pooldavad paljud lastearstid, kliinilised geneetikud ja eetikud sellise operatsiooni edasilükkamist seni, kuni noor inimene on võimeline osalema teadliku otsuse tegemisel.
Vähemalt Põhja-Ameerikas valib vaid väike osa haigestunutest jäsemete pikendamise operatsiooni. Little People of America meditsiininõukogu on avaldanud avalduse jäsemete pikendamise kasutamise kohta.
Rasvumine: Rasvumise ennetamise meetmed peaksid algama varases lapsepõlves. Rasvumise standardsed ravimeetodid peaksid olema akondroplaasiaga inimestel tõhusad, kuigi kalorivajadus on väiksem. [ 26 ]
Edu jälgimiseks tuleks kasutada akondroplaasiale spetsiifilisi standardseid kaalu- ja pikkuse suhtes arvutatud graafikuid. Oluline on märkida, et need kõverad ei ole ideaalsed pikkuse suhtes arvutatud kõverad; need on tuletatud tuhandetest akondroplaasiaga inimeste andmepunktidest.
Kehamassiindeksi (KMI) standardid töötati välja 16-aastastele ja noorematele lastele. [ 27 ] KMI ei ole akondroplaasiaga täiskasvanute jaoks standardiseeritud; võrdlused keskmise pikkuse KMI kõveratega annavad eksitavaid tulemusi. [ 28 ]
Varus-deformatsioon. Soovitatav on iga-aastane ortopeediline järelkontroll kas akondroplaasiaga tuttava arsti või ortopeedilise kirurgi poolt. Kirurgilise sekkumise kriteeriumid on avaldatud.[ 29 ]
Progressiivse sümptomaatilise kõvera olemasolu nõuab suunamist ortopeedi poole. Asümptomaatiline varusdeformatsioon iseenesest ei vaja tavaliselt kirurgilist korrektsiooni. Valida saab mitmesuguste sekkumiste vahel (nt suunatud kasv kaheksa plaadi abil, valgusosteotoomia ja derotatsiooniosteotoomia). Kontrollitud uuringuid, mis võrdleksid ravivõimaluste tulemusi, ei ole.
Küfoos. Akondroplaasiaga imikutel tekib sageli painduv küfoos. Fikseeritud nurgelise küfoosi tekke vältimiseks on olemas protokoll, mis hõlmab painduvate lapsevankrite, kiikede ja kandekottide vältimist. Soovitus mitte toestamata istuda; lapse süles hoidmisel avaldage alati seljale vastusurvet.
- Küfoos paraneb märkimisväärselt või kaob enamikul lastest pärast ortograadse kehahoiaku võtmist ja kõndima hakkamist. [ 30 ]
- Lastel, kellel pärast kere tugevuse suurendamist ja kõndima hakkamist iseenesest ei parane haigus, piisab tavaliselt torakolumbaalse küfoosi püsimise vältimiseks trakside kandmisest.[ 31 ]
- Kui raske küfoos püsib, võib neuroloogiliste tüsistuste vältimiseks olla vajalik selgroooperatsioon.[ 32 ]
Seljaaju stenoos: Seljaaju stenoosi raskete nähtude ja/või sümptomite ilmnemisel on vajalik kiireloomuline suunamine kirurgi spetsialisti juurde.
Tavaliselt soovitatakse laiendatud ja laia laminektoomiat. Protseduuri asjakohasus sõltub stenoosi tasemest (nt rindkere või nimmepiirkond) ja astmest. Patsientidel olid paremad tulemused ja paranenud funktsioon, mida varem neile pärast sümptomite ilmnemist operatsioon tehti [ 33 ].
Vaktsineerimine: Akondroplaasia puhul pole midagi, mis välistaks kõik tavapärased immuniseerimised. Arvestades suurenenud hingamisteede riski, on DTaP, pneumokoki ja gripi vaktsiinid eriti olulised.
Kohanemisvajadused: Lühikese kasvu tõttu võib osutuda vajalikuks keskkonna muutmine. Koolis võib see hõlmata taburette, madalamaid lüliteid, sobiva kõrgusega tualette või muid ligipääsetavuse vahendeid, madalamaid laudu ja jalatugesid toolide ees. Kõik lapsed peaksid hädaolukorras suutma hoonest iseseisvalt lahkuda. Väikesed käed ja nõrgad kõõlused võivad peenmotoorikat raskendada. Sobivate kohanduste hulka kuuluvad väiksema klaviatuuri, raskustega pastakate ja siledamate kirjutuspindade kasutamine. Enamikul lastel peaks olema individuaalse õppekava (IEP) või 504. õppekava.
Sõitmiseks on peaaegu alati vaja pedaalipikendusi. Vajalikuks võib osutuda ka töökoha muutmine, näiteks madalamate laudade, väiksemate klaviatuuride, astmete ja tualeti ligipääsu paigaldamine.
Sotsialiseerumine: Akondroplaasiaga seotud väga märgatava lühikese kasvu tõttu võib mõjutatud inimestel ja nende peredel olla raskusi suhtlemise ja kooliga kohanemisega.
Tugigrupid, näiteks Little People of America, Inc (LPA), saavad aidata peredel neid probleeme lahendada eakaaslaste toetuse, isikliku eeskuju ja sotsiaalse teadlikkuse programmide kaudu.
Teavet tööhõive, hariduse, puuetega inimeste õiguste, lühikeste laste lapsendamise, meditsiiniliste probleemide, sobiva riietuse, abivahendite ja lapsevanemaks olemise kohta saab üleriigilise uudiskirja, seminaride ja töötubade kaudu.
Selle kaasasündinud defekti ravimiseks pole ühtegi ravimit ega ravimivaba ravi.
Kõige sagedamini kasutatakse füsioteraapiat; ravi võib olla vajalik ka hüdrotsefaali (šundi või endoskoopilise ventrikulostoomia abil), rasvumise, [ 34 ] uneapnoe, [ 35 ] keskkõrvapõletiku või seljaaju stenoosi korral.
Mõnes kliinikus tehakse pärast lapse viie- kuni seitsmeaastaseks saamist kirurgilist ravi: sääre-, reie- ja isegi õlaluude luud pikendatakse või deformatsioon korrigeeritakse operatsioonide ja spetsiaalsete ortopeediliste vahendite abil kolmes kuni neljas etapis, millest igaüks kestab kuni 6-12 kuud.
Uurimisjärgus ravi
C-tüüpi natriureetilise peptiidi analoogi manustamine on kliinilistes uuringutes. Esialgsed tulemused on näidanud, et see on hästi talutav ja põhjustab akondroplaasiaga lastel kasvukiiruse suurenemist algtasemega võrreldes ( uuringu keskus ). [ 36 ] Konjugeeritud C-tüüpi natriureetiline peptiid on samuti praegu kliinilistes uuringutes. [ 37 ] Muude kaalutluste hulka kuuluvad türosiinkinaasi inhibeerimine [ 38 ], mekliziin [ 39 ] ja lahustuv rekombinantne inimese FGFR3 peibutis. [ 40 ]
Teavet mitmesuguste haiguste ja seisundite kliiniliste uuringute kohta leiate USA-s veebilehelt clinicaltrials.gov ja Euroopas ELi kliiniliste uuringute registrist.
Ärahoidmine
Ainus ennetav meede on kaasasündinud haiguste sünnieelne diagnoosimine. [ 41 ], [ 42 ]
Prognoos
Kui kaua elavad akondroplaasiaga inimesed? Umbes 10 aastat vähem kui keskmine oodatav eluiga.
Kuna luukoe ja liigeste patoloogilised muutused piiravad enesehooldust ja liikuvust, määratakse selle diagnoosiga lastele puudega inimese staatus. Pikaajaliselt on enamikul patsientidest normaalne prognoos, kuid vanusega suureneb südamehaiguste risk. [ 43 ]