
Pärilik nefriit (Alporti sündroom) lastel
Viimati vaadatud: 23.04.2024

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
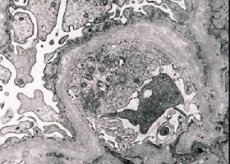
Pärilikud nefriit (Alporti sündroom) - geneetiliselt määratud mitteimmuunsed päritud gloperulopaatia eksponeerimine hematuuria (mõnikord proteinuuria), progresseeruv neerufunktsiooni vähenemise kroonilise neerupuudulikkusega arengut seostatakse tihti kurtus ja nägemispuudega.
LGGuthrie kirjeldas seda haigust esmakordselt 1902. Aastal, kes jälgis perekonda mitu põlvkonda, millest täheldati hematuria. Aastal 1915 kirjeldasid sama AFHurst perekonna liikmed ureemia arengut. Aastal 1927 avastati Alport esmakordselt mitmesuguste hematuria sugulaste kurtus. Eelmise sajandi 50. Aastatel kirjeldati sellises haiguses silmavigastusi. Päriliku hematuriaga patsientidel, neerukude morfoloogiliselt uurides 1972, Hinglais et al. Näitas glomerulaarsete basaalmembraanide ebaühtlane laienemine ja delaminatsioon. 1985. Aastal identifitseeriti päriliku nefriidi geneetiline alus - mutatsioon IV tüüpi kollageeni geenis (Fiengold et al., 1985).
Haiguse geneetilise olemuse uurimine võimaldas järeldada, et päriliku nefriidi fenotüübiliste ilmingute erinevused (koos kuulmiskahjustusega või ilma) tulenevad mutantse geeni ekspressioonitasemest. Seega on praegu kõik kliinilised variandid vaadeldavad ühe haiguse ilminguteks ja termin "pärilik nefriit" on terminiga "Alporti sündroom" sünonüüm.
Epidemioloogiliste uuringute kohaselt esineb pärilik nefriit sagedusega 17 100 000 lapse kohta.
Alporti sündroomi põhjused
Haiguse geneetiline alus on mutatsioon IV tüüpi kollageeni ahela geenis a-5. Seda tüüpi universaalne basaal neeru membraane, cochlear seadme läätse kapslid, võrkkesta ja sarvkesta näidatule uuringud, kus kasutati vastaste monoklonaalsete antikehade kollageeni fraktsioonist. Viimasel ajal osutavad nad võimalusele kasutada DNA sondid päriliku nefriidi prenataalseks diagnoosimiseks.
Rõhutatakse kõigi perekonnaliikmete katsetamise tähtsust DNA-sondide abil mutantse geeni kandjate tuvastamiseks, mis on selle haigusega perede meditsiinilise geneetilise nõustamise läbiviimisel väga tähtis. Kuid kuni 20% peredest ei ole neeruhaigusega sugulastel, mis viitab spontaansete mutatsioonide suurele esinemissagedusele ebanormaalses geenis. Perekondadel pärilik nefriidiga patsientidel on neeruhaigusega inimestel, kuulmislangus ja nägemisega seotud patoloogia; kellel on üks või mitu esivanemat, kuna seotud isikute abielu suurendab tõenäosust, et mõlemad vanemad saavad samad geenid. On kindlaks tehtud autosomaalse dominant-ja autosoomne retsessiivne ja domineeriv, mis on seotud transmissiooniraja X-kromosoomiga.
Lapsed eristavad tõenäolisemalt kolme päriliku nefriidi varianti: Alporti sündroomi, kuulmislangus olevat pärilikku nefriti ja perekonna healoomulist hematuria.
Alporti sündroom - pärilik nefriit koos kuulmiskahjustusega. Selle aluseks on neerude glomerulaatorite basaalmembraani, kõrva ja silma struktuuride ühendatud defekt. Klassikalise Alporti sündroomi geen paikneb X-kromosoomi pika käe lokus 21-22 q. Enamikul juhtudel on see pärilik X-kromosoomiga seotud domineeriv tüüp. Seoses sellega on meestel Alporti sündroom raskem, sest naistel kompenseeritakse mutantset geenifunktsiooni teise, kahjustamata kromosoomi tervel alleelil.
Päriliku nefriidi arengu geneetiline alus on mutatsioonid IV tüüpi kollageeni alfa-ahelate geenides. Ta on tuntud kui kuue-ahelaid IV tüüpi kollageeni G: A5 ja A6 geenide ketid (Sol4A5 ja Sol4A5) asuvad pika käe X kromosoomi 21-22q tsooni; a3- ja a4-ahelate geenid (Co4A3 ja Co4A4) - 2-ndal kromosoomil; a1- ja a2-ahelate geenid (Co4Al ja Co4A2) - 13. Kromosoomis.
Enamikul juhtudel (80-85%) seostatakse X-seotud tüüpi haiguse pärilikkus Co4A5 geeni kahjustusega kustutamise, punktmutatsioonide või splaissimise häirete tõttu. Praegu leitakse rohkem kui 200 mutatsiooni geenist Kol4A5, mis vastutab IV tüüpi kollageeni a5-ahelate sünteesi eest. Sellel pärilikul kujul esineb haigus mõlema sugupoole lastel, kuid poistel on see raskem.
Mutatsioonid geenide Co4A3 ja Co4A4 lookustes, mis on vastutavad IV tüüpi kollageeni a3- ja a4-ahelate sünteesi eest, on autosoomidelt päritud. Uuringu andmetel on autosomaalset domineerivat pärilikku tüüpi täheldatud 16% päriliku nefriidi, autosomaalse retsessiivse juhtudest - 6% patsientidest. Co4A3 ja Co4A4 geenides on umbes 10 mutatsiooni.
Mutatsioonide tulemus on IV tüüpi kollageeni kokkupaneku protsesside rikkumine, mis põhjustab selle struktuuri häireid. Kollageeni tüüp IV on üks glomerulaarse basaalse membraani, košulaarse aparatuuri ja silma läätse peamistest komponentidest, mille patoloogia ilmneb päriliku nefriidi kliinikus.
IV tüüpi kollageeni, osa glomerulaarfiltratsiooni basaalmembraan, koosneb põhiliselt kahest ahelast a1 (IV) ja üks a2 ahela (IV) ja sisaldab ka a3, a4, a5 ahela. Enamasti kui X-seotud pärandi Sol4A5 mutatsioon kaasneb vähene a3, a4- ja a6 A5 ahelaid IV tüüpi kollageeni struktuuri ja mitmeid O1 ja a2 ahelaid glomerulaarse basaalmembraani suureneb. Selle nähtuse mehhanism on ebaselge, eeldatakse, et põhjus on transkriptsioonilised muutused mRNA-s.
Vähene a3, A4- ja A5 ketid struktuuri tüüp IV kollageeni basaalmembraani glomeruli tulemusi hõrenemine ja haprus algstaadiumis Alport sündroom, mis avaldub kliiniliselt kõige hematuuria (mõnikord hematuuria või proteinuuria ainult proteinuuria), kuulmislangus ja lenticonus. Edasine haiguse kulgu põhjustab paksenemine ja häireid basaalmembraani läbilaskvus hilisstaadiumites haiguse, kusjuures kasv nendes tüüpi kollageeni V ja VI, mis väljendub suurendamist proteinuuria ja neerufunktsiooni vähenemine.
Päriliku nefriidi aluseks oleva mutatsiooni olemus määrab suuresti selle fenotüübilise ilmingu. Kui X-kromosoomi deletsiooni samaaegsete mutatsioonide ja Sol4A6 Sol4A5 eest vastutavate geenide sünteesi A5 ja A6 chains IV tüüpi kollageeni, mis on kombineeritud Alporti sündroom leiomyomatosis söögitoru ja suguelunditele. Vastavalt uuringud Sol4A5 geenimutatsioonid seostatakse deletsioon on tähistatud suurte raskusest patoloogilist protsessi, kombinatsiooni koos neerukahjustus Ekstrarenaalse ilmingud ja varases tekkes krooniline neerupuudulikkus, võrreldes stochechnoy mutatsioon Selle geeni.
Morfoloogiliselt näitab elektronmikroskoopia glomerulaarsete basaalmembraanide (eriti lamina densa) hõrenemist ja delaminatsiooni ning elektrooniliselt tihedate graanulite olemasolu. Glomerulaadi kahjustus võib sama patsiendil olla mittenõueteta, alates mesangiooni minimaalsest fookuskahjustusest kuni glomeruloskleroosi. Gloterüliit Alporti sündroomis on alati immuun-negatiivne, mis eristab seda glomerulonefriidist. Iseloomulikud on kanali atroofia, lümfiohistüotsüütide infiltratsiooni areng, "vahtrakkude" esinemine lipiidide - lipofaagi lisamisega. Haiguse progresseerumisel avastatakse basaallampide membraanide paksenemine ja märgatav hävimine.
Ilmuvad teatavad muutused immuunsüsteemi seisundis. Patsiendid, kellel on pärilik nefriit väiksemal määral Ig A- ja kalduvus tõusu kontsentratsioon veres IgM, IgG tase võib suurendada algetappidel haiguse ja taandarengu hilisemates etappides. Võib-olla on IgM ja G kontsentratsiooni suurenemine mingi kompenseeriv reaktsioon vastuseks IgA defitsiidile.
T-lümfotsüütide süsteemi funktsionaalne aktiivsus on vähenenud; See tähistas selektiivse reduktsiooniga B-lümfotsüüdid, sünteesi eest vastutava Ig A, purustatud fagotsütoossete immuunsuse lingile, mille peamiseks põhjuseks jaotus protsesside kemotaksise ja rakusisese seedimist neutrofiilide
Uuringus neerubiopsia patsientidel Alporti sündroom elektronmikroskoobiga, ultrastrukturaalsed täheldatud muutused glomerulaarfiltratsiooni basaalmembraani: hõrenemine ja lõikamise mustrid rikkumise glomerulaarfiltratsiooni alusmembraanides muutusega selle paksus ja ebaühtlane kontuurid. Päriliku nefriidi varases staadiumis määrab defekt glomerulaarsete basaalmembraanide hõrenemise ja haprususe.
Glomerulaarmembraanide leotamine on soodsam märk ja tüdrukute hulgas on see sagedasem. Päriliku nefriidi püsiv elektronmikroskoopiline funktsioon on basaalmembraani lõhestamine ja selle hävitamise raskusaste korreleerub protsessi tõsidusega.

Alporti sündroomi sümptomid lastel
Esimesed kolm eluaastat on sagedamini esinenud Alporti sündroomi esimesi sümptomeid isoleeritud kuseteinisündroomi kujul. Enamikul juhtudest avastatakse haigus juhuslikult. Kuseelundi sündroom ilmneb lapse ennetava uurimise käigus enne lasteasutuse või ARVI sisenemist. Kui ARVI-l tekib patoloogiat uriinis. Päriliku nefriidi korral, erinevalt omandatud glomerulonefriidist, ei ole varjatud perioodi.
Haiguse algfaasis on lapse heaolu vähe, iseloomulik omadus on kusete sündroomi püsivus ja püsivus. Üks peamisi sümptomeid on erineval määral hematuria, mida täheldati 100% juhtudest. Hematuria taseme tõus on täheldatud hingamisteede infektsioonide, füüsilise koormuse või pärast profülaktilist vaktsineerimist. Proteinuuria enamikul juhtudel ei ületa 1 g päevas, haiguse alguses võib olla ebastabiilne, kuna protsessi käigus suureneb proteinuuria. Perioodiliselt võib uriiniga setetel esineda leukotsütopeenia, millel on lümfotsüütide ülekaal, mis on seotud interstitsiaalsete muutuste arenguga.
Hiljem on rikutud neerude osalisi funktsioone, patsiendi üldise seisundi halvenemist: joobeseisundit, lihaste nõrkust, arteriaalset hüpotensiooni, sageli kuulmispuudega (eriti poistega), mõnikord nägemise halvenemist. Mürgistus esineb halvasti, väsimus, peavalu. Haiguse algfaasis tuvastatakse enamikul juhtudest kuulmiskaod ainult audiograafia abil. Alporti sündroomi kuulmiskaod võivad tekkida erinevatel lapsepõlves, kuid sagedamini kuulmiskaod diagnoositakse vanuses 6-10 aastat. Laste kuulmiskaod algavad suurel sagedusel, saavutades märkimisväärselt õhu ja luu juhtimise, läbides heli juhtivuse ja heli vastuvõttev kuulmiskaotus. Kuulmiskaod võivad olla üks esimesi haiguse sümptomeid, mis võivad eelneda uriinisündroomile.
20% -l juhtudest on Alporti sündroomiga patsientidel muutused silmas. Kõige tavalisemad läätsed: spherofokiya, lentikonus eesmine, tagumine või segatud, mitmesugused kataraktid. Alporti sündroomiga peredel on lühiajaline lühiajaline esinemissagedus. Mitmed teadlased on pidevalt nende perekondade tähistada kahepoolse perimakulyarnye muudatusi hele valkjas või kollakas granulatsioonide on kollaskeha. Nad peavad seda sümptomit pideva sümptomina, millel on Alporti sündroomi puhul kõrge diagnostilist väärtust. C. S. Chugh et al. (1993) oftalmoloogilisteks uurimusest selgus Alporti sündroomiga patsientidel vähenenud nägemisteravus 66,7% juhtudest, ettepoole lenticonus - 37,8%, laikude võrkkestal - in 22,2%, katarakt - 20%, keratoconus - 6 , 7%.
Mõnedel päriliku nefriidi lastel, eriti neerupuudulikkuse tekkimisel, on märkimisväärne hilistumine füüsilises arengus. Kuna neerupuudulikkuse progresseerumisel tekib hüpertensioon. Lastel esineb seda sagedamini noorukieas ja vanemates vanuserühmas.
Iseloomulik on mitmesuguste (rohkem kui 5-7) sidekoe düsembriogeneesi häirete päriliku nefriidi esinemine patsientidel. Hulgas sidekoe stigma patsientidel kõige sagedasem silmade hüpertelorism, kõrge suulagi, malocclusion, ebanormaalne kuju kõrvad, kumerust väikese sõrme tema käed, "sandalevidnaya lõhe" jalgadel. Pärilike nefriit iseloomustab ühetaolisuse dizembriogeneza häbimärgistamine perekonnas, samuti suure sagedusega nende turustamiseks katseisikutest sugulased, mille kaudu haigust edastab.
Varasematel haigusetappe paljastas isoleeritud vähendamine osalise neerutalitluse: transport aminohapete, elektrolüüdid, kontsentratsiooni funktsioone Acidogenesis, edasiste muudatuste funktsionaalse seisundi nii proksimaalse ja distaalse nefronite ja on oma olemuselt Ühendatud osalise häired. Glomerulaarfiltratsiooni vähendamine toimub hiljem, sagedamini noorukieasperioodil. Kui pärilik nefriit areneb, tekib aneemia.
Seega pärilike nefriit erineb lavastuses haiguse: esimene latentse etapil või varjatud kliiniliste sümptomitega avaldub minimaalseid muutusi põie sündroom seejärel toimub järkjärgult dekompensatsioonita protsessi vähenemisega neerufunktsiooni kliinilise avaldumisega sümptomid (mürgistus, jõuetus, arengu hilinemist, anemizatsiya). Kliinilised sümptomid ilmnevad tavaliselt, olenemata põletikulise reaktsiooni kihistumisest.
Pärilik nefriit võib ilmneda erinevates vanuseperioodides, mis sõltub geeni toimest, mis teatud aja jooksul on represseeritud.
Klassifikatsioon
Päriliku nefriidi kolm varianti on olemas
- I variant - ilmneb kliiniliselt hematuuria, kuulmislanguse ja silmakahjustusega nefriidiga. Nefriidi kulg on progresseeruv CRF-i arenguga. Pärandi tüüp on domineeriv, seotud X-kromosoomiga. Morfoloogiliselt on häiritud basaalse membraani struktuur, selle hõrenemine ja lõhustamine.
- II variant - kliiniliselt väljendub nefriit koos hematuriaga ilma kuulmiskahjustuseta. Nefriidi kulg on progresseeruv koos kroonilise neerupuudulikkuse arenguga. Pärandi tüüp on domineeriv, seotud X-kromosoomiga. Morfoloogiliselt leitakse glomerulaarsete kapillaaride (eriti laminaadensa) basaalmembraani hõrenemine.
- III valik - healoomuline perekondlik hematuria. Kurss on soodne, krooniline neerupuudulikkus ei arene. Päriliku liik on autosoomne valitsev või autosoomne retsessiivne. Autosomaalsel retsessiivne pärilikku tüüpi naistel on haiguse raskem käik.
Alporti sündroomi diagnoosimine
Kavandatakse järgmised kriteeriumid:
- vähemalt kahe neerupõletikuga patsiendi esinemine igas perekonnas;
- hematuria kui nefropaatia juhtiv sümptom proovides;
- vähemalt ühel pereliikmel on kuulmiskaotus;
- kroonilise neerupuudulikkuse areng ühes sugulasel ja rohkem.
Diagnoosimisel erinevaid pärilik ja kaasasündinud haiguste oluline koht kuulub integreeritud lähenemisviisi kontroll ja ennekõike pöörata tähelepanu andmete valmistamisel saadud lapse põlvnemist. Diagnoos sündroom Alporti mõjuvaks juhtudel, kus patsient 3 otsa 4 tüüpilised omadused: esinemisega perekonna hematuuria ja krooniline neerupuudulikkus, esinemine patsiendi sensorineuraalsete kuulmiskadu, patoloogiate avastamis- elektrontiheduse mikroskoopilist iseloomustamiseks biopsia märke lõikamisreaktsioonides glomerulaarfiltratsiooni basaalmembraani koos muutustega tema paksuse ja ebaühtlane kontuur.
Patsiendi uurimine peaks hõlmama uuringute kliinilisi geneetilisi meetodeid; haiguse anamneesi suunatud uuring; patsiendi üldine uurimine, võttes arvesse diagnostilisi kriteeriume. Hüvitusetapis saab patoloogiat püüda vaid keskendudes sellistele sündroomidele nagu pärilike komplikatsioonide, hüpotensiooni, düsembriogeneesi mitmesuguse häbimärgistuse, muutused kusete sündroomis. Dekompenseerunud estrarenalnyh võib põhjustada selliseid sümptomeid nagu raske joove, jõuetus, aeglustatud füüsilist arengut anemizatsiya avaldub ja võimendamise koos järkjärguline vähenemine neerufunktsiooni. Enamuses neerufunktsiooni langusega patsientidest täheldatakse happelise ja aminogeneesi funktsiooni halvenemist; 50% patsientidest märgib neerude sekretoorse funktsiooni märkimisväärset langust; uriini optilise tiheduse kõikumiste piiramine; filtreerimise rütmi rikkumine ja seejärel glomerulaarfiltratsiooni vähenemine. Kroonilise neerupuudulikkuse tase diagnoositakse patsientidel, kellel on vereseerumis 3-6 kuud ja rohkem karbamiidi taseme (rohkem kui 0,35 g / l), glomerulaarfiltratsiooni vähenemine 25% -ni normist.
Diferentsiaaldiagnoosimine pärilikud nefriit peab toimuma peamiselt omandatud vormiga hematuric glomerulonefriit. On pälvinud järjest teravamaks glomerulonefriit algavad 2-3 nädala jooksul pärast eelmist infektsioon, Ekstrarenaalse funktsioone, sealhulgas hüpertensiooni esimese päeva (päriliku nefriit vastupidi, hüpotensioon), glomerulaarfiltratsiooni kiiruse vähenemine haiguse alguses ei ole rikutud osaline torukujulise funktsioone, arvestades nagu pärilikult, nad on kohal. Omandatud glomerulonefriit toimub raskema hematuuria ja proteinuuria, suurenenud ESR. Diagnostiline väärtus on tüüpilised muutused glomerulaarfiltratsiooni basaalmembraani iseloomulik pärilik nefriit.
Diferentsiaaldiagnoosimine düsmetaboolsed nefropaatia läbi Kroonilise neerupuudulikkusega perekonnas eristab kliiniliselt monotüüpia neeruhaigus ja võib ulatuda nefropaatia püelonefriit urolitiaasile. Lapsed kannatavad sageli kõhuvalu ja urineerimise perioodil, uriini setetes - oksalaadis.
Kui teil on pärilik nefriit kahtlustatav, tuleb patsiendile saata diagnoosi selgitamiseks spetsialiseeritud nefroloogia osakonnas.
Mida tuleb uurida?
Kuidas uurida?
Millised testid on vajalikud?
Kellega ühendust võtta?
Alporti sündroomi ravi
Režiim võimaldab piirata suurt füüsilist koormust, püsida värskes õhus. Toitumine on kõrge kvaliteediga, piisava hulga kõrgekvaliteetsetest valkudest, rasvadest ja süsivesikutest, võttes arvesse neerufunktsiooni. Suur tähtsus on krooniliste infektsioonipõletike tuvastamine ja rehabilitatsioon. Ravimitest kasutatakse ATP, karboksülaasi, püridoksiini (kuni 50 mg päevas), karnitiinkloridiini. Kursused toimuvad 2-3 korda aastas. Kui hematuria on ette nähtud fütoteraapia - nõges, nõges, mustad tuhad, põõsas.
Välisriigis ja kodumaises kirjanduses on teatatud prednisolooniravist ja tsütostaatikumide kasutamisest. Kuid mõju on raske hinnata.
Kroonilise neerupuudulikkuse korral kasutatakse hemodialüüsi ja neeru siirdamist.
Päriliku nefriidi spetsiifilise (efektiivse patogeense) ravi ei ole olemas meetodeid. Kõik meditsiinilised meetmed on suunatud neerufunktsiooni vähendamise vältimiseks ja aeglustamiseks.
Toit peaks olema tasakaalustatud ja kõrge kalorsusega, võttes arvesse neerude funktsionaalset seisundit. Lapse toitumisfunktsiooni häirete puudumisel peaks laps olema piisav valkude, rasvade ja süsivesikute sisaldus. Neerupuudulikkuse nähtude ilmnemisel tuleks piirata proteiini, kaltsiumi ja fosfori süsivesikute sisaldust, mis viivitab kroonilise neerupuudulikkuse arengut.
Füüsiline stress peaks olema piiratud, lastel soovitatakse hoiduda sportimisest.
Vältige kontakti nakkustega patsientidega, vähendage ägedate hingamisteede infektsioonide tekkimise riski. On vaja puhastada kroonilise infektsiooni fookusi. Päriliku nefriidi korral ei tehta ennetavaid vaktsineerimisi, vaktsineerimine on võimalik ainult vastavalt epidemioloogilistele näidustustele.
Päriliku nefriidi hormonaalsed ja immunosupressiivsed ravimid on ebaefektiivsed. On teatatud teatud positiivsest mõjust (proteinuuria taseme langus ja haiguse progresseerumise aeglustumine) koos tsüklosporiin A ja AKE inhibiitorite pikaajalise kasutamisega.
Patsientide ravis, kes kasutavad aineid, mis parandavad ainevahetust:
- püridoksiin - 2-3 mg / kg / päevas 3 jagatud annusena 4 nädala jooksul;
- kokarboksilaas - 50 mg intramuskulaarselt igal teisel päeval, ainult 10-15 süstet;
- ATP - 1 ml intramuskulaarselt iga päev, 10-15 süstet;
- A-vitamiin - 1000 U / aastas / päev 1 vastuvõtt 2 nädalat;
- E-vitamiin - 1 mg / kg päevas 1 vastuvõtt 2 nädala jooksul.
Selline ravi parandab patsientide üldist seisundit, vähendab toruja düsfunktsiooni ja seda manustatakse 3 korda aastas.
Immunomodulaatorina võib kasutada levamisooli - 2 mg / kg / päevas 2-3 korda nädalas, mille vahele jäävad 3-4 päeva annused.
Teadlastele avaldab hüperbaarne oksügenisatsioon positiivset mõju hematuria raskusele ja neerufunktsiooni häirele.
Päriliku nefriidi ravimise kõige efektiivsem meetod on õigeaegne neeru siirdamine. Juhul kui seda ei täheldatud siirdamisoperatsiooni retsidiivis, väheses koguses (umbes 5%) võib nefriit arengut siirdatud neeru, seostatakse antigeene glomerulaarfiltratsiooni basaalmembraani.
Paljutõotav ala on sünnieelne diagnoos ja geenitehnoloogia teraapia. Loomkatsed näitavad, et tüüpiline IV kollageeni a-ahela sünteesimine neerukoes põhjustab tavapäraste geenide kõrge efektiivsuse, mille järel on täheldatud normaalsete kollageenstruktuuride sünteesi.
Prognoos
Päriliku nefriidi prognoos on alati tõsine.
Päriliku nefriidi voolu prognostiliselt ebasoodsad kriteeriumid on:
- meeste sugu;
- kroonilise neerupuudulikkuse varajane areng pereliikmetel;
- proteinuuria (üle 1 g päevas);
- glomerulaarsete basaalmembraanide paksenemine vastavalt mikroskoobile;
- kuulmisnärvi neuritis;
- deletsioon geenis Co4A5.
Parema hematuria prognoos on soodsam.
Использованная литература