Artikli meditsiiniline ekspert

Uued väljaanded
Neuroblastoom lastel: põhjused, diagnoosimine, ravi
Viimati vaadatud: 04.07.2025

Kõik iLive'i sisu vaadatakse meditsiiniliselt läbi või seda kontrollitakse, et tagada võimalikult suur faktiline täpsus.
Meil on ranged allhanke juhised ja link ainult mainekate meediakanalite, akadeemiliste teadusasutuste ja võimaluse korral meditsiiniliselt vastastikuste eksperthinnangutega. Pange tähele, et sulgudes ([1], [2] jne) olevad numbrid on nende uuringute linkideks.
Kui tunnete, et mõni meie sisu on ebatäpne, aegunud või muul viisil küsitav, valige see ja vajutage Ctrl + Enter.
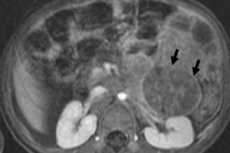
Laste onkoloogias on üks levinumaid ekstrakraniaalseid kasvajaid lastel neuroblastoom, mis on närviharja neuroblastide ehk sümpaatilise närvisüsteemi embrüonaalsete (ebaküpsete) närvirakkude embrüonaalne pahaloomuline kasvaja.
Epidemioloogia
Rahvusvahelise Neuroblastoomi Riskirühma (INRG) statistika kohaselt moodustab neuroblastoom umbes 8% kõigist laste onkoloogilistest haigustest kogu maailmas ja on levimuselt kolmas pärast leukeemiat ja ajukasvajaid.
Teiste andmete kohaselt moodustab neuroblastoom umbes 28% kõigist imikute vähijuhtudest. Rohkem kui kolmandik neuroblastoomi juhtudest diagnoositakse alla üheaastastel lastel; diagnoosimise keskmine vanus on 19–22 kuud. Üle 90% diagnoositud juhtudest esineb kahe- kuni viieaastastel lastel (peamiselt poistel); esinemissageduse tipphetk on kahe- kuni kolmeaastasel lapsel ja üle viieaastaste laste hulgas on juhtumeid alla 10%.
Põhjused neuroblastoomid
Neuroblastoomi põhjuste uurimisel on teadlased jõudnud järeldusele, et see kasvaja lastel tekib embrüogeneesi või varase postnataalse arengu ajal esinevate juhuslike geneetiliste mutatsioonide tõttu. Kuid mis neid geenimuutusi põhjustab, pole teada, kuna teratogeensete keskkonnategurite mõju pole kindlaks tehtud.
Need kasvajad võivad esineda kõikjal, sealhulgas mediastinumis, kaelas, kõhus, neerupealistes, neerudes, selgroos ja vaagnas.
Harvadel juhtudel võib imikute neuroblastoom olla seotud päriliku mutatsiooniga. Eelkõige mutatsiooniga membraanvalgu CD246 geenis 2. kromosoomis – ensüümi türosiinkinaasi ALK geenis, mis tagab rakkudevahelise kommunikatsiooni ja mängib olulist rolli närvisüsteemi toimimises; valgu PHOX2B geenis (4. kromosoomis), mis osaleb närvirakkude küpsemises.
Neuroblastoom võib olla seotud ka lapsepõlves esineva 1. tüüpi neurofibromatoosiga,Beckwith-Wiedemanni sündroomiga ja hüperinsulinemilise hüpoglükeemiaga (nesidioblastoosne pankreatiit).
Riskitegurid
Tänapäeval peetakse pärilikkust lastel neuroblastoomi tekke riskiteguriteks - selle kasvaja esinemist perekonnaanamneesis, samuti kaasasündinud anomaaliaid, mis on seotud geenimutatsioonidega emakasisese arengu ajal. See kehtib eriti mitmete kasvajate tekke kohta erinevates organites.
Teadlased ei ole tuvastanud ühtegi eksogeenset tegurit, mis suurendaks selle kasvaja riski.
Pathogenesis
Neuroblastoomide arengu mehhanism on põhjustatud närviharja rakkude diferentseerumise ja küpsemise häiretest – need on kahepoolsed rakuliinid, mis moodustuvad inimese embrüo ektodermaalsest idukihist närvitoru servades. Need rakud migreeruvad (liiguvad) ja diferentseeruvad mitut tüüpi rakkudeks: sensoorsed ja autonoomsed neuronid, neuroendokriinsed rakud ja neerupealise medulla rakud, kraniofakiaalse kõhre ja luude rakud, samuti pigmendirakud.
Neuroblastoomi korral migreerunud neuroblastid ei küpse, vaid jätkavad kasvu ja jagunemist, moodustades kasvaja. Ja selle moodustumise patogenees on seotud järgmiste geenimutatsioonidega:
- embrüo närviharja rakkudes RBTN1 valku kodeeriva LMO1 geeni kromosoomijärjestuse osa dubleerimisega või segmentide dubleerimisega 11. kromosoomis;
- inimese närvisüsteemi tüvirakkude proliferatsiooni kontrolliva DUF1220 valku kodeeriva NBPF10 geeni koopiaarvu muutusega kromosoomis 1q21.1. Need häired põhjustavad kas selle kromosoomi dubleerimist või deletsiooni – DNA osa puudumist;
- muutustega kasvaja supressorgeenis ATRX (kromosoomis Xq21.1);
- N-Myc transkriptsioonifaktori geeni täiendavate koopiate (amplifikatsiooni) olemasolul 2. kromosoomis, mis kodeerib ühte transkriptsioonifaktorit (DNA-d siduv valk), mis reguleerib teiste geenide aktiivsust ja kontrollib eellasrakkude proliferatsiooni loote kudede ja organite moodustumiseks vajalike valkude moodustumise ajal. Selle geeni amplifikatsioon muudab selle onkogeeniks, mis provotseerib rakutsükli häireid, suurenenud rakkude proliferatsiooni ja kasvajate moodustumist.
Sümptomid neuroblastoomid
Neuroblastoomi esimesed nähud on mittespetsiifilised ja võivad hõlmata isutuskaotust (ja kaalulangust), väsimust söötmisel, palavikku ja liigesevalu.
Kliinilised sümptomid sõltuvad primaarse kasvaja asukohast ja metastaaside olemasolust (mis esinevad 60–73% juhtudest).
Väga sageli lokaliseerub primaarne neuroblastoom neerupealise medullas, millel on närvirakkudega sarnane päritolu. Alla üheaastastel lastel diagnoositakse neerupealise neuroblastoom 35–40% juhtudest. Selle sümptomiteks on kõhuvalu, palavik, kaalulangus, luuvalu, aneemia või samaaegne Pepperi sündroom: difuusne maksakahjustus koos raske hepatomegaalia ja respiratoorse distressi sündroomiga.
Retroperitoneaalne neuroblastoom ehk retroperitoneaalne neuroblastoom lastel hakkab kasvades suruma põiele või sooltele, mis võib põhjustada probleeme urineerimise või roojamisega, jalgade turset (poistel paisub munandikott).
Laste mediastiinumi neuroblastoom (mediastiinumi neuroblastoom) surub sageli ülemisele õõnesveenile ja see võib põhjustada näo, kaela, käte ja rindkere ülaosa turset (nahk muutub sinakaspunaseks koos nahaaluste sõlmedega). Tekivad köha ja vilistav hingamine, hingamisprobleemid (õhupuudus) või neelamisprobleemid (düsfaagia); suurenenud lümfisõlmed on täheldatud kaelal, rangluu kohal ja kaenlaalustes.
Kasvajarakkude levik luuüdisse põhjustab aneemiat, trombotsütopeeniat ja leukopeeniat, millega kaasneb kalduvus verejooksule.
Ja periorbitaalse piirkonna metastaaside korral tekivad silmade ümber tumedad ringid või verevalumid. Selline kasvaja võib põhjustada ka peavalu ja pearinglust, eksoftalmiat (silmamunade punnitamist) ning närvilõpmete kokkusurumise tõttu - silmalaugude rippumist (ptoos) ja pupillide suuruse vähenemist (mioos).
Kõhuõõne neuroblastoom ehk laste kõhuõõne neuroblastoom põhjustab kõhus palpeeritavate tihendite teket, kõhu paisumist, isutust, kõhukinnisust ja vererõhu tõusu. Seljaajule või närvijuurele suruv kasvaja võib põhjustada jäsemete tuimust ja nõrkust, võimetust seista, roomata või kõndida. Kui luud on kahjustatud, võib tekkida luuvalu.
Lümfisõlmede kahjustusega kõhuõõne 3.-4. staadiumi kasvaja korral võivad kasvajarakud siseneda neeruparenhüümi ja seejärel areneb lastel ulatuslik neeru neuroblastoom, mis viib selle funktsioonide häidumiseni.
Etapid
- 1. staadiumi neuroblastoom on primaarne kasvaja, mis on lokaliseeritud ja isoleeritud ühes kehapiirkonnas; mõlema poole lümfisõlmed ei ole mõjutatud.
- Neuroblastoomi 2. staadium. 2A staadiumis piirdub primaarne kasvaja ühe piirkonnaga, kuid on suur; kahepoolsed lümfisõlmed ei ole kahjustatud. 2B staadiumis on metastaasid leitud kehapoole lümfisõlmedes, kus kasvaja asub.
- Neuroblastoomi 3. staadium: primaarne kasvaja läbib seljaaju või keha keskjoont, lümfisõlmedes leidub ühe- või kahepoolseid metastaase.
- Neuroblastoomi 4. staadium: kasvaja on levinud kaugematesse lümfisõlmedesse, luuüdisse, luudesse, maksa või teistesse organitesse. 4S staadium määratakse alla üheaastastel lastel, kellel on lokaliseeritud primaarne kasvaja, mis on levinud nahka, maksa või luuüdisse.
Rahvusvaheline neuroblastoomi riski staadiumisüsteem (INRGSS)
INRGSS kasutab pildipõhiselt määratletud riskitegureid (IDRF), mis on pildiuuringutel nähtavad tegurid, mis võivad tähendada, et kasvajat on raskem eemaldada.
INRGSS jagab neuroblastoomid neljaks staadiumiks:
- L1: Kasvaja ei ole levinud algkohast ega ole kasvanud elutähtsateks struktuurideks. See piirdub ühe kehaosaga, näiteks kaela, rindkere või kõhuga.
- L2: Kasvaja ei ole levinud (metastaseerunud) kaugele kohast, kus see algas (näiteks võib see olla kasvanud kõhu vasakust küljest rindkere vasakule küljele), kuid sellel on vähemalt üks IDRF.
- M: Kasvaja on metastaseerunud keha kaugemasse ossa (välja arvatud MS-staadiumis kasvajad).
- MS: Metastaatiline haigus alla 18 kuu vanustel lastel, mille puhul vähk on levinud ainult nahka, maksa ja/või luuüdisse.
Tüsistused ja tagajärjed
Neuroblastoomi iseloomustavad sellised tüsistused ja tagajärjed nagu:
- levik (metastaasid) lümfisõlmedesse, luuüdisse, maksa, nahka ja luudesse;
- seljaaju kokkusurumine (mis võib põhjustada valu ja viia halvatuseni);
- paraneoplastilise sündroomi teke (kasvaja poolt eritatavate teatud kemikaalide, samuti selle rakkude poolt ekspresseeritud antigeeni disialogangliosiidi GD2 toimel), mis avaldub kiirete tahtmatute silmaliigutuste, koordinatsioonihäirete, lihaskrampide ja kõhulahtisuse näol;
- ägenemised pärast esmase ravi lõpetamist (nagu näitab kliiniline praktika, on kõrge riskiga neuroblastoomidel 50% juhtudest retsidiiv).
Diagnostika neuroblastoomid
Lapse kahtlustatava neuroblastoomi diagnoosimiseks on vaja läbivaatust, laboratoorseid uuringuid ja pildistamist.
Vere- ja uriinianalüüsid tehakse katehhoolamiinide (norepinefriin ja dopamiin) ning homovanilliin- või vanillüülmandelhapete (moodustuvad nende hormoonide metabolismi käigus) määramiseks; neurospetsiifilise enolaasi vereanalüüs, vereseerumi ensüümse immunosorbentanalüüsi (ELISA) meetod ja luuüdi analüüs (proov võetakse aspiratsioonipunktsiooni teel). Mutatsioonide määramiseks tehakse DNA-test ja kasvajakoe tsütomorfoloogiliseks uurimiseks tehakse biopsia.
Pärast biopsiaproovide võtmist saadetakse need laborisse, kus patoloog (arst, kellel on eriväljaõpe vähirakkude tuvastamisel) uurib neid mikroskoobi all. Proovidega tehakse sageli ka spetsiaalseid laborikatseid, et teha kindlaks, kas kasvaja on neuroblastoom.
Kui tegemist on neuroblastoomiga, võivad laborikatsed aidata kindlaks teha ka seda, kui kiiresti kasvaja võib kasvada või levida, ning millised ravimeetodid võivad kõige paremini toimida.
Instrumentaalne diagnostika visualiseerib neoplasmi ultraheli, röntgeni, MRI või KT, PET-i abil 18F-fluorodeoksüglükoosi lisamisega või MIBG-skaneerimise - stsintigraafia abil metajodobensüülguanidiiniga. [ 1 ]
Diferentseeritud diagnoos
Diferentsiaaldiagnoos hõlmab healoomulist ganglioneuroomi, ganglioneuroblastoomi, rabdomüosarkoomi, nefroblastoomi.
Kellega ühendust võtta?
Ravi neuroblastoomid
Neuroblastoomi ravi sõltub patsiendi riskigrupist (kasvajaprotsessi staadiumist), kasvaja lokaliseerimisest, kasvajarakkude genoomsetest omadustest ja lapse vanusest. See võib hõlmata jälgimist, kirurgiat, keemiaravi, kiiritusravi, immunoteraapiat ja vereloome tüvirakkude siirdamist.
Nagu iga vähikeemiaravi, manustatakse ka lastel neuroblastoomi neoadjuvantset ehk adjuvantset (pre- või postoperatiivset) keemiaravi kuuridena: ravimit manustatakse mitu päeva järjest, millele järgneb paus, et organism saaks taastuda. Tsükleid korratakse tavaliselt iga kolme kuni nelja nädala järel.
Kasutatakse järgmisi ravimeid (ja nende kombinatsioone): tsüklofosfamiid, tsisplatiin või karboplatiin, doksorubitsiin (adriamütsiin), vinkristiin, etoposiid.
Keemiaravi ravimite levinud kõrvaltoimete hulka kuuluvad juuste väljalangemine, isutus, väsimus, iiveldus ja oksendamine, suuhaavandid, kõhulahtisus või kõhukinnisus. Keemiaravi võib kahjustada luuüdi ja põhjustada vererakkude arvu vähenemist.
Sihipärane immunoteraapia (mis on suunatud kasvaja antigeenile GD2) kasutab monoklonaalsete antikehade (anti-GD2 MAb) rühma kuuluvaid ravimeid dinutuksimabi (Unituxin) ja naksitamabi. Neid manustatakse intravenoosselt pikaajalise infusioonina kombinatsioonis granulotsüütide-makrofaagide kolooniat stimuleeriva faktoriga (tsütokiin GM-CSF) ja interleukiin-2-ga.
Nende ravimite kõrvaltoimete hulka kuuluvad valu (sageli väga tugev), vererõhu langus, südame löögisageduse tõus, õhupuudus (koos võimaliku hingamisteede tursega), palavik, iiveldus, oksendamine ja kõhulahtisus, muutused vere rakulises ja mineraalses koostises.
Vähi kordumise riski vähendamiseks pärast suurtes annustes keemiaravi ja tüvirakkude siirdamist ravitakse kõrge riskiga neuroblastoomiga lapsi süsteemsete retinoididega, 13-cis-retinoehappega (isotretinoiin). [ 2 ]
Neuroblastoomi kirurgiline ravi – kasvaja eemaldamine, näiteks neerupealise neuroblastoomi avatud adrenalektoomia või laparoskoopiline resektsioon; lümfektoomia (kahjustatud lümfisõlmede eemaldamine) jne [ 3 ]
Kõrge riskiga neuroblastoomi korral võib kasutada kiiritusravi.[4 ]
Ärahoidmine
Arvestades neuroblastoomi põhjuseid lastel, võib ainsaks ennetavaks meetmeks olla geneetiline nõustamine raseduse planeerimisel. Kuid tuleb meeles pidada, et see kasvaja on seotud pärilike mutatsioonidega ainult 1-2% juhtudest.
Prognoos
Infantiilsel neuroblastoomil on võime spontaanselt taanduda.
Prognostilised markerid
- Kõrge riskiga kasvajad, samuti neuroblastoom igas vanuses ja igas staadiumis (välja arvatud 4S staadium) lastel – koos N-MYC geeni suurenenud ekspressiooni ja N-Myc onkogeeni amplifikatsiooniga – on ebasoodsa prognoosiga, mis mõjutab oodatavat eluiga.
- Kui kasvajarakkudel puuduvad 1. või 11. kromosoomi teatud osad (tuntud kui 1p või 11q deletsioonid), on prognoos halvem. Samuti on halvem prognoos seotud 17. kromosoomi lisaosaga (17q juurdekasv).
- Suure hulga DNA-ga neuroblastoomirakkudel on parem prognoos, eriti alla 2-aastastel lastel.
- Neuroblastoomidel, millel on rohkem neurotrofiini retseptoreid, eriti närvikasvufaktori retseptorit TrkA, on parem prognoos.
Ellujäämine lapsepõlve onkoloogiagrupi (COG) riskigrupi järgi
- Madala riskiga rühm: Madala riskiga rühma kuuluvate laste 5-aastane elulemus on üle 95%.
- Keskmise riskiga rühm: Keskmise riskiga rühma kuuluvate laste 5-aastane elulemus on 90–95%.
- Kõrge riskiga rühm: Kõrge riskiga laste 5-aastane elulemus on umbes 50%.
Umbes 15% laste vähisurmadest on põhjustatud neuroblastoomist. Selle kõrge riskiga pahaloomulise kasvaja pikaajalise ellujäämise tõenäosus ei ületa 40%. Üldine viieaastane elulemus on 67–74%, ühe- kuni nelja-aastaste laste puhul 43% ja esimesel eluaastal diagnoositud neuroblastoomi korral üle 80%.
Использованная литература